Introduction and Definition of Pierre Robin Sequence (PRS)
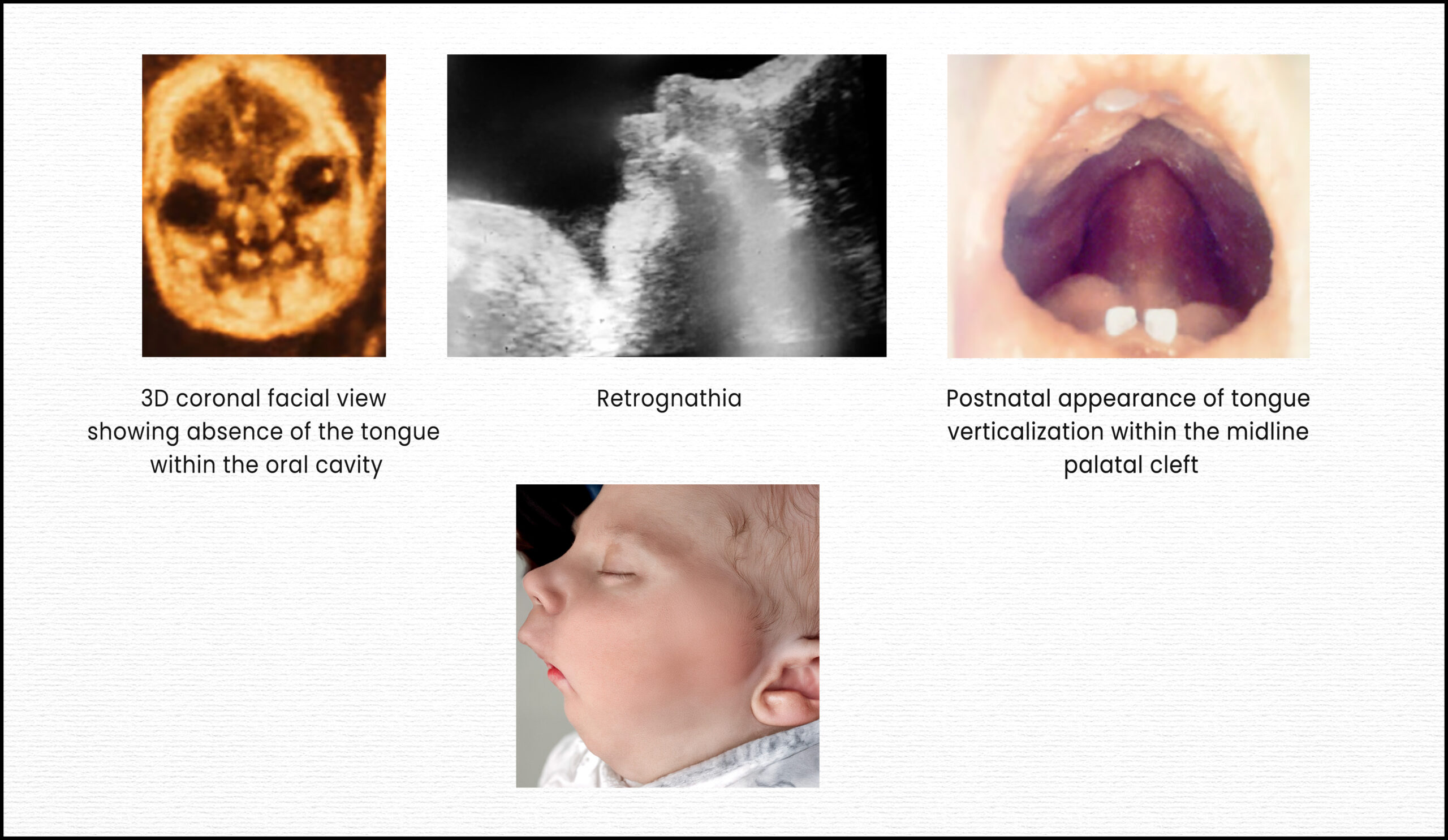
Pierre Robin sequence (PRS) is a rare congenital anomaly characterized by a classic triad of symptoms: micrognathia (undersized jaw), glossoptosis (posterior displacement or falling back of the tongue), and upper airway obstruction. A U-shaped cleft palate is frequently, though not always, associated with this triad. This condition is termed a “sequence” because micrognathia is considered the primary developmental disturbance, which then leads to glossoptosis, and subsequently, airway obstruction. The retropositioning of the tongue can also inhibit the fusion of the palatal shelves, often resulting in the characteristic U-shaped cleft palate.
PRS is a heterogeneous entity, meaning its presentation can vary widely. It can occur as nonsyndromic PRS (nsPRS), where only the core triad (with or without cleft palate) is present, or as syndromic PRS (sPRS), where it is associated with other congenital anomalies or genetic conditions. A distinct classification, PRS Plus, is used for nonsyndromic cases that exhibit additional malformations. The incidence of PRS is reported to range from 1 in 8,500 to 1 in 30,000 live births, with the highest reported incidence in the USA at 1 per 3,120 live births. Syndromic cases account for approximately 50% of all PRS occurrences, and around 34 different syndromes have been linked to PRS.
The clinical significance of PRS lies primarily in the potential for life-threatening respiratory obstruction and feeding difficulties, which contribute to a high mortality rate reported between 1.7% and 65%. Mortality rates are notably higher in children with associated syndromes, particularly those involving cardiac and central nervous system comorbidities. Consequently, the overarching priority in PRS management is to maintain a viable upper respiratory tract.
The Role of Genetic Understanding
Understanding the genetic mutations associated with PRS is crucial for several reasons. Such knowledge can facilitate early confirmatory diagnosis and guide the formulation of appropriate, tailored treatment plans. Furthermore, genetic mutations linked to PRS can be detected prenatally, sometimes as early as the first trimester of pregnancy. This early detection empowers parents to make informed decisions regarding the continuation of the pregnancy, especially given the potential severity of the syndrome. Despite its importance, a comprehensive collection of literature on genetic mutations in PRS has been lacking.
Methodology of the Systematic Review
To address this gap, the authors conducted a systematic review adhering to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement. Their search spanned Web of Science, PubMed, and Scopus databases, utilizing the keywords “Pierre Robin syndrome/sequence AND gene mutation”. The review specifically included original research, case reports, and case series written in English that detailed genetic mutations in PRS, while excluding reviews, conference abstracts, animal models, and articles lacking sufficient genetic information.
Out of an initial 208 articles, 39 met the stringent eligibility criteria for qualitative analysis after a rigorous screening process that involved identifying duplicates and irrelevant studies. The high kappa coefficient values (0.98 and 0.96 for the two screening steps) indicate strong reliability between the reviewers. In terms of bias assessment for the included case series and reports, 34 studies were categorized as having a low risk of bias (meeting 80-100% of quality criteria), while one study had a moderate risk of bias (meeting 60% of criteria).
Key Findings of the Systematic Review
The 39 selected articles collectively reported on 324 cases of PRS. The distribution of these cases highlighted the prevalence of associated conditions:
- Syndromic PRS (sPRS): 182 cases (56%).
- Nonsyndromic PRS (nsPRS): 120 cases (37%).
- PRS Plus: 22 cases (6.8%), referring to nsPRS with additional malformations.
The review identified Stickler syndrome as the most common associated syndrome, appearing in 30 cases (16% of sPRS). Other frequently associated conditions included chromosomal abnormalities (17 cases, 9%), Richieri-Costa-Pereira syndrome (15 cases, 8%), Catel-Manzke Syndrome (10 cases, 5.5%), and TARP Syndrome (6 cases, 3%). It was noted that for 46 sPRS cases (25%), the specific associated syndrome was not mentioned or evaluated.
Various diagnostic techniques were employed to assess genetic mutations, most commonly FISH and G-banding. Other methods included exome sequencing, polymerase chain reaction, microarray, Sanger sequencing, array-CGH, next-generation sequencing, conformation-sensitive gel electrophoresis, and expression profiling.
Crucially, 100 out of the 324 cases (30.9%) had identified genetic mutations. The types of mutations observed varied, with deletions being the most frequent (47%), followed by translocations (20%), duplications (12%), and single nucleotide polymorphisms (9%). Abnormal karyotypes were present in 34 of these cases.
The SOX9 gene was found to be the most commonly mutated gene, identified in 22 cases. SOX9 is critical for cartilage development, and its mutation was linked to isolated PRS, campomelic dysplasia, acampomelic dysplasia, and PRS Plus cases. Specifically, loss of function or haploinsufficiency of SOX9 is associated with lethal skeletal malformations and syndromic PRS. In contrast, disruptions upstream or downstream of the SOX9 gene, which involve highly conserved noncoding cis-regulatory elements, are linked to milder abnormalities and nonsyndromic PRS.
Other genes implicated in isolated PRS included KCNJ2 (12 cases), BMPR1B (5 cases), COL11A1 (6 cases), COL11A2 (1 case), and COL2A1 (4 cases). Chromosome 2 mutations were also found in isolated PRS. For syndromic PRS, the genetic mutations were largely specific to the associated syndrome, such as TGDS in Catel-Manzke syndrome, RBM10 in TARP syndrome, and SNRPB in cerebro-costo-mandibular syndrome. In PRS Plus cases, although SOX9 mutations were common, other mutations like BMP2, NF2, MN1, MAP2K6, BMP4, OTX2, and KCNJ2 were also observed.
Regarding familial inheritance, only 21 of the 39 studies assessed genetic mutations in family members. Several instances of familial inheritance were reported, including autosomal dominant and recessive patterns, as well as maternal inheritance, across various PRS classifications and associated syndromes.
Limitations of the Reviewed Literature and Recommendations
The systematic review highlighted several limitations within the existing literature on PRS genetic mutations. A significant concern was the heterogeneity of the studies; only 5 were cohort studies, with the majority being case reports or case series, often involving very small sample sizes. These small sample sizes hinder robust statistical evaluation of factors like disease progression and treatment response. Furthermore, a notable deficiency was the limited assessment of familial inheritance patterns, with less than half of the studies investigating family members. There remains an ambiguity as to whether the identified genetic mutations are the sole cause of PRS or merely associated findings. The absence of gene ontology and pathway enrichment analysis in most studies (only one study performed this) further limits the understanding of the exact etiopathogenesis and the clinical implications of these mutations.
To advance the understanding and management of PRS, the authors strongly recommend several key actions for future research:
- Conduct large-scale, multicenter cohort studies to provide sufficient statistical power for evaluating disease progression and treatment outcomes.
- Implement a standardized protocol for assessing genetic mutations across all cases to minimize bias stemming from variations in diagnostic modalities.
- Prioritize pedigree analysis to comprehensively assess potential familial inheritance patterns.
- Integrate gene ontology and pathway enrichment analysis to gain deeper insights into the etiopathogenesis, mutation severity, and clinical manifestations, thereby aiding in more precise treatment planning.
In conclusion, while significant strides have been made in identifying genetic mutations associated with PRS, a more coordinated and large-scale research effort is imperative to fully elucidate its genetic basis and improve patient care.