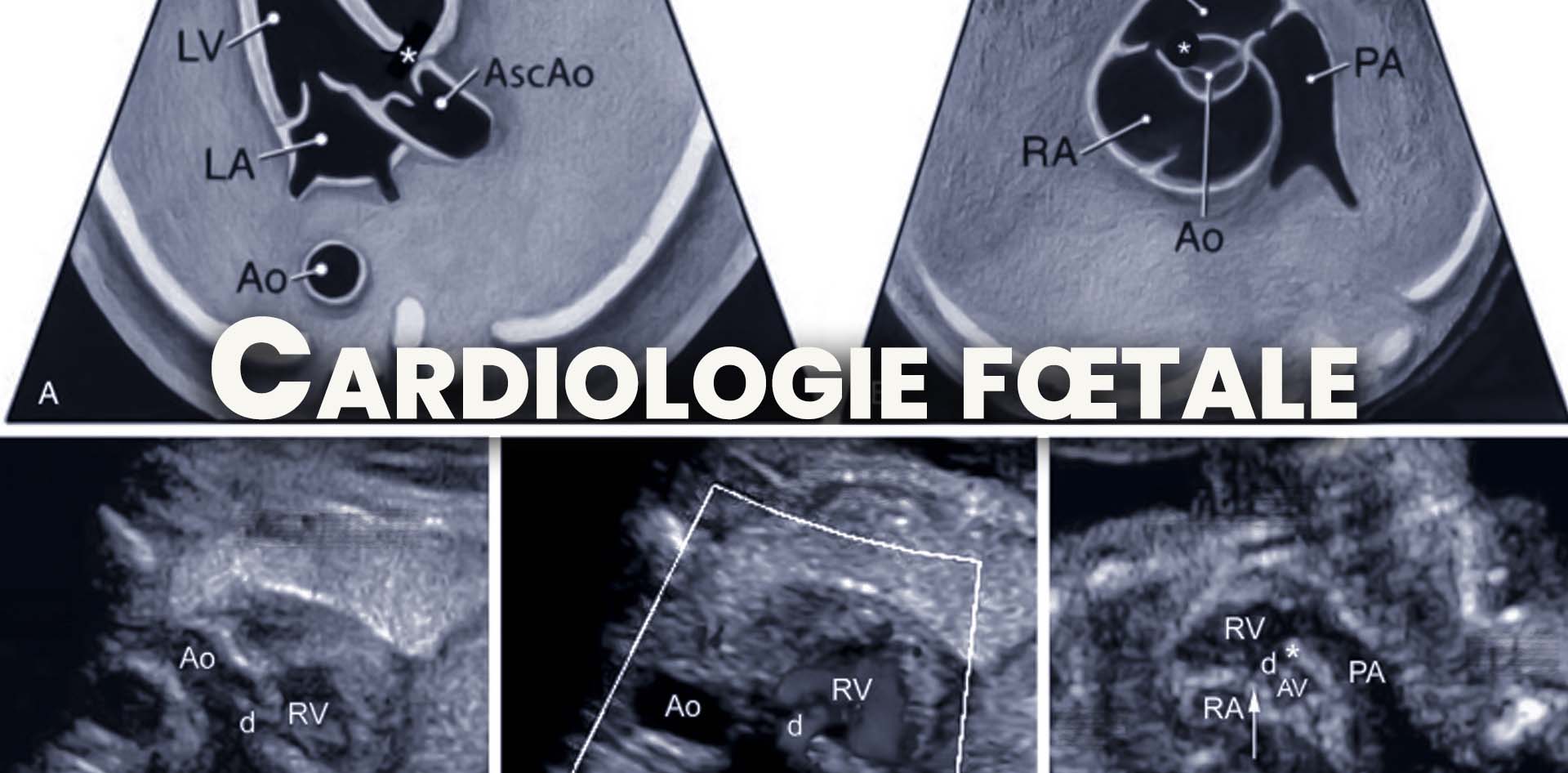
Les cardiopathies congénitales (CC) constituent les malformations structurelles les plus fréquentes, touchant environ 1 % des naissances vivantes. Autrefois limitée à un diagnostic anatomique souvent partiel, la cardiologie fœtale s’est transformée en une discipline d’excellence, plaçant désormais le « fœtus comme patient » au cœur d’une prise en charge multidisciplinaire. Cette maturité repose sur un double socle : une compréhension approfondie de la physiologie circulatoire et l’accès à une technologie d’imagerie toujours plus performante.
L’évolution technologique a radicalement modifié le dépistage. Si la coupe des quatre cavités était autrefois le standard, ne détectant que 50 % des anomalies majeures, l’adoption systématique d’un protocole incluant cinq coupes axiales a permis de porter ce taux de détection à près de 90 %. De plus, les progrès de la résolution d’image permettent désormais une détection précoce dès 11 à 14 semaines de gestation, offrant un délai précieux pour le conseil génétique et la planification thérapeutique.
Au-delà de l’échographie conventionnelle, la discipline s’est enrichie de modalités avancées. L’IRM cardiaque fœtale (CMR) permet aujourd’hui de caractériser des malformations complexes lorsque l’échographie est limitée par la position fœtale ou l’habitus maternel. Parallèlement, la magnétocardiographie fœtale (fMCG) s’impose comme l’outil le plus précis pour l’analyse des troubles du rythme et de la repolarisation, permettant de diagnostiquer des syndromes du QT long ou des torsades de pointes in utero avec une précision comparable à l’ECG postnatal.
Cette puissance technologique s’accompagne d’une meilleure connaissance de l’histoire naturelle des lésions. Nous savons aujourd’hui que certaines cardiopathies sont évolutives. Une sténose aortique critique peut progresser in utero vers un syndrome d’hypoplasie du cœur gauche, une cascade que des interventions telles que la valvuloplastie aortique fœtale tentent désormais de briser pour préserver une circulation biventriculaire à la naissance.
Un autre signe de la maturité de la discipline est l’intégration de la protection cérébrale dans les enjeux cardiologiques. L’imagerie par résonance magnétique a révélé que les perturbations de l’hémodynamique cardiaque impactent directement le métabolisme et la maturation du cerveau fœtal. Cette vision globale permet d’affiner le pronostic neuro-développemental à long terme, bien au-delà de la simple survie périnatale.
La décision clinique s’est également structurée autour de modèles de stratification des risques. L’organisation des naissances par niveaux de soins (LOC 1 à 4) garantit que les nouveau-nés porteurs de lésions critiques reçoivent des soins de réanimation ou des interventions salvatrices, comme la septostomie atriale, immédiatement après l’accouchement. Cette logistique de précision est le fruit d’une collaboration étroite au sein d’équipes de cardio-obstétrique regroupant obstétriciens, cardiologues pédiatres, généticiens et néonatologistes.
En conclusion, la cardiologie fœtale a dépassé le stade de la simple curiosité diagnostique pour devenir une science de la décision. Grâce à des outils de plus en plus performants et une expertise clinique affinée, elle offre aujourd’hui aux familles une information précise et des perspectives thérapeutiques concrètes, optimisant ainsi l’avenir de chaque enfant né avec une anomalie cardiaque.
Approche systématique devant une anomalie cardiaque fœtale
Pr Yves Ville
Les cardiopathies congénitales (CC) touchent environ 1 % des naissances vivantes et constituent une cause majeure de mortalité néonatale.
- Dépistage et Indications de l’Échocardiographie Fœtale
Le dépistage s’adresse à la fois aux populations à bas risque (routine) et à haut risque. Bien que 90 % des CC surviennent sans facteur de risque identifiable, certaines indications imposent une échocardiographie fœtale spécialisée :
- Facteurs maternels : Diabète prégestationnel, maladies auto-immunes (anticorps anti-Ro/SSA et anti-La/SSB), ou exposition à des tératogènes (lithium, rétinoïdes).
- Facteurs familiaux : Antécédents de CC chez un parent ou un germain.
- Facteurs fœtaux : Anomalie suspectée lors d’une échographie de routine, clarté nucale (CN) augmentée (≥3,5 mm), anomalies extracardiaques, arythmies ou suspicion d’anomalie chromosomique.
- Protocoles de Diagnostic et Techniques d’Imagerie
L’échocardiographie fœtale, réalisée idéalement entre 18 et 22 semaines de gestation, est l’outil de référence. Elle repose sur une analyse segmentaire séquentielle de l’anatomie cardiaque.
L’examen de base s’est élargi de la simple coupe des quatre cavités (4C) pour inclure les voies d’éjection ventriculaires et la coupe des trois vaisseaux et de la trachée (3VT). La méthode des cinq coupes axiales, balayant du thorax supérieur jusqu’à l’abdomen, permet de détecter jusqu’à 90 % des CC majeures.
L’utilisation du Doppler couleur et pulsé est indispensable pour évaluer la fonction valvulaire et les flux vasculaires. L’imagerie avancée, telle que l’IRM cardiaque fœtale ou la magnétocardiographie (fMCG), peut compléter le diagnostic, notamment pour les anomalies des gros vaisseaux ou les arythmies complexes. Un diagnostic précoce dès 11-14 semaines est possible par voie transabdominale ou transvaginale pour les grossesses à haut risque.
III. Évaluation du Rythme et de la Fonction Cardiaque
L’analyse systématique inclut la mesure de la fréquence cardiaque (normale : 120-160 bpm) et la recherche d’arythmies. Les tachycardies soutenues ($>$ 160-180 bpm) ou les bradycardies ($<$ 120 bpm) nécessitent une expertise pour en déterminer le mécanisme (ex: bloc auriculo-ventriculaire complet) et guider le traitement.
L’évaluation de la fonction cardiaque utilise le Score de Profil Cardiovasculaire (CVP). Ce score sur 10 points évalue cinq catégories :
- Signes d’hydrops (épanchements, œdème).
- Taille du cœur (rapport cardiothoracique).
- Fonction ventriculaire (fraction de raccourcissement).
- Flux Doppler veineux (canal d’Arantius, veine ombilicale).
- Flux Doppler artériel (artère ombilicale).
Un score CVP $\le$ 7 indique un risque accru de compromis cardiovasculaire et de mortalité périnatale.
- Recherche d’Anomalies Associées
La découverte d’une anomalie cardiaque doit déclencher une recherche systématique d’autres malformations :
- Anomalies extracardiaques : Présentes dans 20 % à 50 % des cas de CC fœtales (anomalies gastro-intestinales, neurologiques ou génito-urinaires).
- Anomalies génétiques : Le risque d’aneuploïdie (trisomie 21, 18, 13) ou de microdélétions (22q11.2) est élevé. Le conseil génétique et le caryotype ou l’analyse par puce à ADN (CMA) sont recommandés. Le séquençage de l’exome peut augmenter le rendement diagnostique en cas d’anomalies multiples.
- Prise en Charge et Planification de la Naissance
La gestion nécessite une équipe multidisciplinaire incluant cardiologues pédiatriques, obstétriciens, néonatologistes et chirurgiens.
- Suivi sériel : Des échographies régulières (toutes les 2 à 4 semaines selon la lésion) permettent de surveiller la progression de la maladie (ex: évolution d’une sténose aortique vers un syndrome d’hypoplasie du cœur gauche).
- Intervention fœtale : Dans des cas sélectionnés, comme la sténose aortique critique avec foramen ovale restrictif, une valvuloplastie in utero peut être envisagée pour tenter de préserver une circulation biventriculaire.
- Stratification du risque à l’accouchement : Le lieu et le mode d’accouchement sont planifiés selon quatre niveaux de soins (LOC) :
- LOC 1 & 2 : Risque faible à modéré, surveillance néonatale standard ou début de prostaglandines.
- LOC 3 & 4 : CC critiques (ex: transposition des gros vaisseaux avec septum intact) nécessitant une intervention immédiate en salle de naissance (septostomie atriale) ou un transfert urgent vers un centre de chirurgie cardiaque.
Conclusion
Une approche systématique, intégrant un diagnostic anatomique précis, une évaluation fonctionnelle par le score CVP et un dépistage génétique, transforme le pronostic des CC. Cette stratégie permet d’optimiser les soins prénataux, d’offrir un conseil éclairé aux parents et d’assurer une transition sécurisée vers la vie postnatale dans des centres spécialisés.
Génétique et détection des anomalies
uhfuhf
Cette synthèse explore l’approche systématique de la détection des anomalies, des marqueurs échographiques précoces jusqu’aux outils génomiques les plus avancés.
- Le dépistage par marqueurs échographiques au premier trimestre
La détection des anomalies commence souvent dès la fin du premier trimestre (11 à 13 semaines + 6 jours). L’outil de dépistage principal est la mesure de la clarté nucale (CN), une accumulation transitoire de liquide dans les tissus sous-cutanés de la nuque fœtale.
- La clarté nucale (CN) : Elle est considérée comme augmentée lorsqu’elle dépasse le 95e ou le 99e percentile, ou qu’elle est $\ge$ 3,5 mm. Une CN augmentée est un marqueur puissant d’aneuploïdies (comme la trisomie 21) mais aussi de CC structurelles, même chez les fœtus dont le caryotype est normal. Le risque de CC est directement proportionnel à l’épaisseur de la CN.
- Marqueurs Doppler : L’évaluation du flux dans le canal d’Arantius (ductus venosus) et la recherche d’une régurgitation tricuspide sont des compléments essentiels. Un flux inversé de l’onde ‘a’ dans le canal d’Arantius ou une régurgitation tricuspide augmentent significativement la probabilité d’une anomalie cardiaque majeure. L’utilisation combinée de ces marqueurs permet de détecter plus de 50 % des CC majeures dès le premier trimestre.
- Les anomalies chromosomiques et structurelles classiques
La découverte d’une anomalie structurelle, qu’elle soit cardiaque ou extracardiaque, impose une recherche génétique. Environ 15 % à 40 % des fœtus porteurs de CC présentent une anomalie chromosomique.
- Aneuploïdies : Les trisomies 21 (syndrome de Down), 18 (syndrome d’Edwards) et 13 (syndrome de Patau), ainsi que la monosomie X (syndrome de Turner), constituent la majorité des cas détectés par caryotype classique ou QF-PCR. Par exemple, 40 % à 50 % des enfants trisomiques 21 ont une CC, souvent un canal atrioventriculaire (CAV).
- Anomalies de nombre de copies (CNV) : L’analyse par puce à ADN (CMA) permet de détecter des microdélétions et microduplications invisibles au caryotype standard. La microdélétion 22q11.2 (syndrome de DiGeorge) est la plus fréquente, associée à 75 % de CC, notamment des anomalies conotroncales comme la tétralogie de Fallot ou l’interruption de l’arche aortique. Le CMA augmente le rendement diagnostique d’environ 4 % à 10 % par rapport au caryotype seul.
III. L’ère du séquençage de nouvelle génération (NGS)
Lorsque les tests conventionnels (Caryotype, CMA) sont négatifs, le recours au séquençage de l’exome (ES) ou du génome (WGS) est désormais recommandé, en particulier pour les anomalies multiples.
- Rendement diagnostique de l’exome : Une méta-analyse majeure montre que l’ES apporte un rendement diagnostique supplémentaire moyen de 31 % chez les fœtus présentant des anomalies structurelles et un CMA normal. Ce rendement est optimisé par une sélection rigoureuse des cas par des équipes multidisciplinaires.
- Variabilité selon le phénotype : Le succès de l’ES dépend du système organique touché. Il est particulièrement élevé pour les anomalies squelettiques (53 %) et neurologiques (13,8 %), tandis qu’il est plus faible pour une CN augmentée isolée (environ 2 %). Pour les CC isolées, le rendement additionnel de l’ES est d’environ 11,5 %.
- Mutations de novo vs héréditaires : Les CC sporadiques sont souvent dues à des mutations de novo (nouvelles variantes chez le fœtus, absentes chez les parents). Ces mutations touchent fréquemment des gènes codant pour des modificateurs de la chromatine ou des voies de signalisation comme Notch. À l’inverse, les anomalies de latéralité (hétérotaxie) montrent un enrichissement en mutations récessives héritées.
- Syndromes monogéniques et CC
De nombreux syndromes génétiques complexes associent des CC à des anomalies extracardiaques :
- Syndrome de Noonan : Lié à des mutations de la voie RAS/MAPK (ex: PTPN11), il se manifeste souvent par une sténose pulmonaire et une hypertrophie cardiaque.
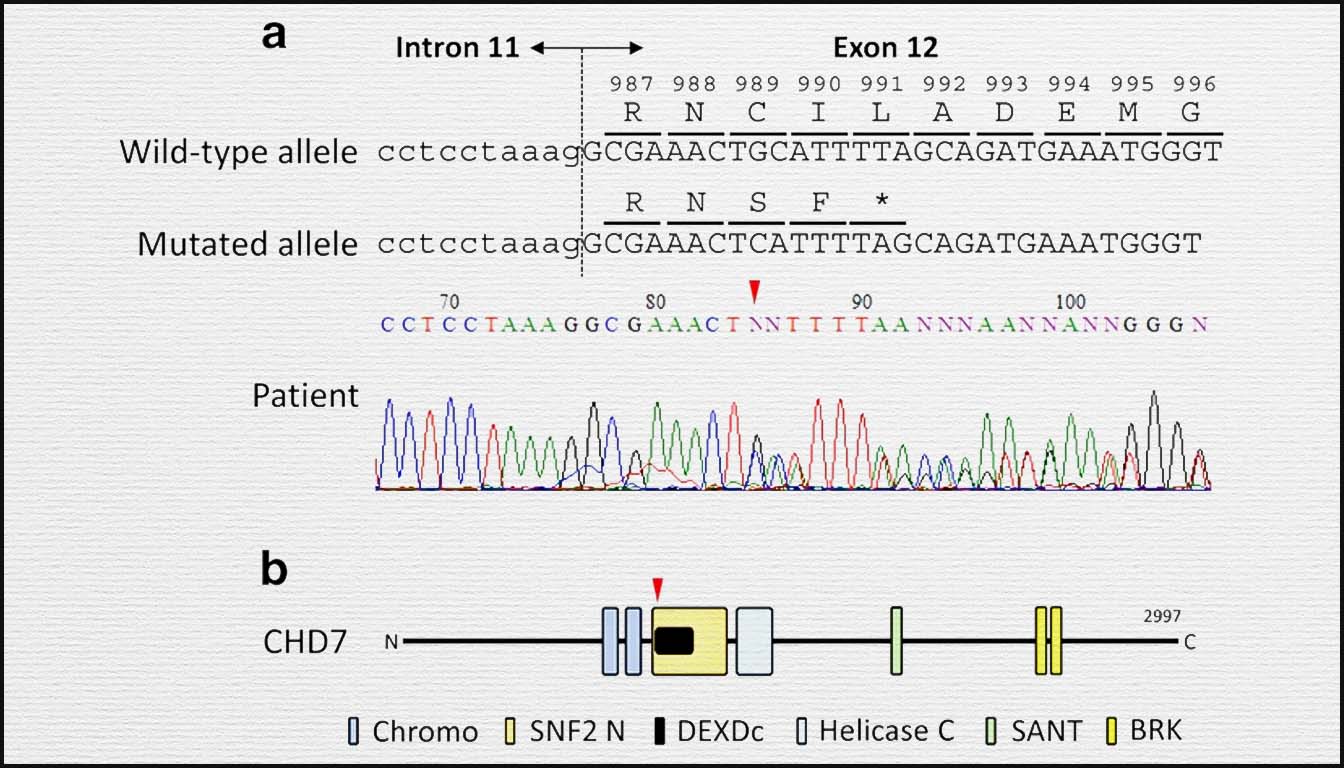
- Syndrome CHARGE : Causé par des mutations du gène CHD7, il inclut des anomalies cardiaques, une atrésie des choanes et des anomalies de l’oreille.
- Syndrome de Kabuki : Souvent associé à une coarctation de l’aorte et des malformations septales.
- L’artère subclaviaire droite aberrante (ARSA) : Bien qu’elle puisse être un signe d’appel, l’ARSA isolée est considérée comme une variante bénigne et n’est généralement pas associée à un risque accru d’anomalies chromosomiques en l’absence d’autres marqueurs.
- Impact clinique et prise en charge périnatale
L’identification d’une cause génétique est cruciale pour plusieurs raisons :
- Pronostic et décision : Elle permet d’informer les parents sur le pronostic neuro-développemental et fonctionnel, facilitant les décisions concernant la poursuite de la grossesse ou l’interruption médicale (IMG).
- Planification de la naissance : Les CC majeures nécessitent un accouchement dans un centre de niveau 3 ou 4 capable de réaliser des interventions immédiates comme la septostomie atriale ou la chirurgie cardiaque néonatale.
- Conseil génétique : La connaissance du mode de transmission (dominant, récessif ou de novo) permet d’évaluer précisément le risque de récurrence pour les grossesses futures (allant de négligeable pour une mutation de novo à 25-50 % pour les formes mendéliennes).
Conclusion
La détection systématique des anomalies fœtales repose sur une intégration étroite entre l’échographie experte du premier et du deuxième trimestres et une exploration génétique graduelle. Si le caryotype et le CMA restent les premières étapes, le séquençage de l’exome est devenu un outil indispensable pour résoudre les cas complexes. Cette approche globale permet non seulement d’améliorer la survie néonatale par une prise en charge ciblée, mais aussi d’offrir aux familles une compréhension claire des origines de l’anomalie et des perspectives pour l’avenir.
Signes échographiques clés des cardiopathies fœtales
Pr Pierre Mace
Cette synthèse détaille les signes clés organisés par plans anatomiques, marqueurs fonctionnels et stades de la grossesse.
- La Coupe des Quatre Cavités (4C) : Fondements de l’Analyse
La coupe 4C est la base de l’examen, permettant de détecter environ 50 % des CC majeures. Un examen normal doit confirmer plusieurs éléments critiques :
- Situs et Position : Le cœur doit être situé majoritairement dans l’hémithorax gauche avec l’apex pointant à gauche selon un axe de 45° ± 20°. Un axe anormal est un signe d’appel fort pour des anomalies des voies d’éjection ou des anomalies chromosomiques.
- Taille Cardiaque : Le cœur doit normalement occuper environ un tiers de la surface thoracique. Un rapport cardiothoracique augmenté (cardiomégalie) est souvent le premier signe d’une insuffisance valvulaire sévère ou d’une dysfonction myocardique.
- Symétrie et Structure : Les deux oreillettes et les deux ventricules doivent être de taille approximativement égale en milieu de gestation. Une disproportion ventriculaire (VD > VG) est un signe suspect pour une coarctation de l’aorte ou un syndrome d’hypoplasie du cœur gauche (SHCG).
- La Croix du Cœur : L’intégrité de la croix du cœur, avec le décalage normal des valves auriculo-ventriculaires (la tricuspide s’insérant plus près de l’apex que la mitrale), doit être visible. L’absence de ce décalage est caractéristique d’un canal atrioventriculaire (CAV).
- Les Voies d’Éjection et la Coupe des Trois Vaisseaux (3VT)
L’ajout des voies d’éjection et de la coupe 3VT permet d’augmenter le taux de détection jusqu’à 90 %.
- Croisement des Gros Vaisseaux : Dans un cœur normal, l’aorte et l’artère pulmonaire se croisent à angle droit à leur origine. La visualisation de vaisseaux parallèles sortant des ventricules est le signe pathognomonique de la transposition des gros vaisseaux (TGV).
- Le “V” de la Coupe 3VT : Normalement, l’isthme aortique et le canal artériel convergent à gauche de la trachée pour former une image en “V”.
- Une image en “U” entourant la trachée indique une artère aortique droite ou un anneau vasculaire.
- Un vaisseau unique dans le médiastin peut signaler un troncus arteriosus ou une atrésie pulmonaire/aortique sévère.
- Une asymétrie de taille entre les deux branches du “V” (isthme aortique étroit) suggère une coarctation de l’aorte.
III. Apports du Doppler Couleur et Pulsé
Le Doppler est indispensable pour valider l’anatomie et évaluer la fonction.
- Flux Rétrograde : Un flux inversé (bleu au lieu de rouge ou vice-versa) dans l’isthme aortique ou le canal artériel est un signe de cardiopathie ducto-dépendante, comme une atrésie aortique ou pulmonaire.
- Régurgitations Valvulaires : Une régurgitation tricuspide holosystolique est un marqueur important de CC structurelle et d’aneuploïdie, particulièrement au premier trimestre. Une régurgitation mitrale sévère peut accompagner une sténose aortique critique.
- Flux Veineux : Un flux inversé de l’onde ‘a’ dans le canal d’Arantius (ductus venosus) ou des pulsations dans la veine ombilicale sont des signes de dysfonction diastolique ou d’augmentation de la pression auriculaire droite.
- Signes Clés par Pathologie Majeure
Les sources permettent d’identifier des combinaisons de signes spécifiques :
- Tétralogie de Fallot (TOF) : Présence d’une communication interventricularie (CIV) large, d’une aorte “à cheval” sur le septum et d’une artère pulmonaire de petite taille.
- SHCG : Ventricule gauche hypoplasique (parfois invisible), atrésie ou sténose de la mitrale, et flux rétrograde dans l’arche aortique en 3VT.
- Anomalie d’Ebstein : Déplacement apical sévère de la tricuspide, oreillette droite géante et régurgitation tricuspide massive.
- Canal Atrioventriculaire (CAV) : Valve unique commune (image en “Y” au Doppler), défaut des septums auriculaire et ventriculaire, et perte de l’offset normal des valves.
- Dépistage Précoce et Évaluation Globale
Dès 11 à 13 semaines + 6 jours, certains signes prédisent une CC même en l’absence de diagnostic anatomique complet. Une clarté nucale (CN) $\ge$ 3,5 mm est associée à un risque significativement accru de CC structurelle. L’utilisation combinée de la CN, du Doppler du canal d’Arantius et de la recherche d’une régurgitation tricuspide permet de suspecter plus de 50 % des CC majeures dès ce stade.
Enfin, le Score de Profil Cardiovasculaire (CVP) synthétise ces signes sur 10 points (hydrops, taille du cœur, fonction ventriculaire, Doppler artériel et veineux). Un score $\le$ 7 est un indicateur de mauvais pronostic et de risque de mortalité périnatale.
Conclusion
La détection efficace des CC fœtales repose sur la reconnaissance de ruptures de symétrie, d’anomalies de flux Doppler et de déviations d’axes vasculaires. La standardisation des protocoles incluant systématiquement la 4C, les voies d’éjection et la 3VT est la clé pour minimiser les faux négatifs et optimiser la prise en charge néonatale.
Doppler et détection précoce des anomalies cardiaques
Dr PIERRE mace
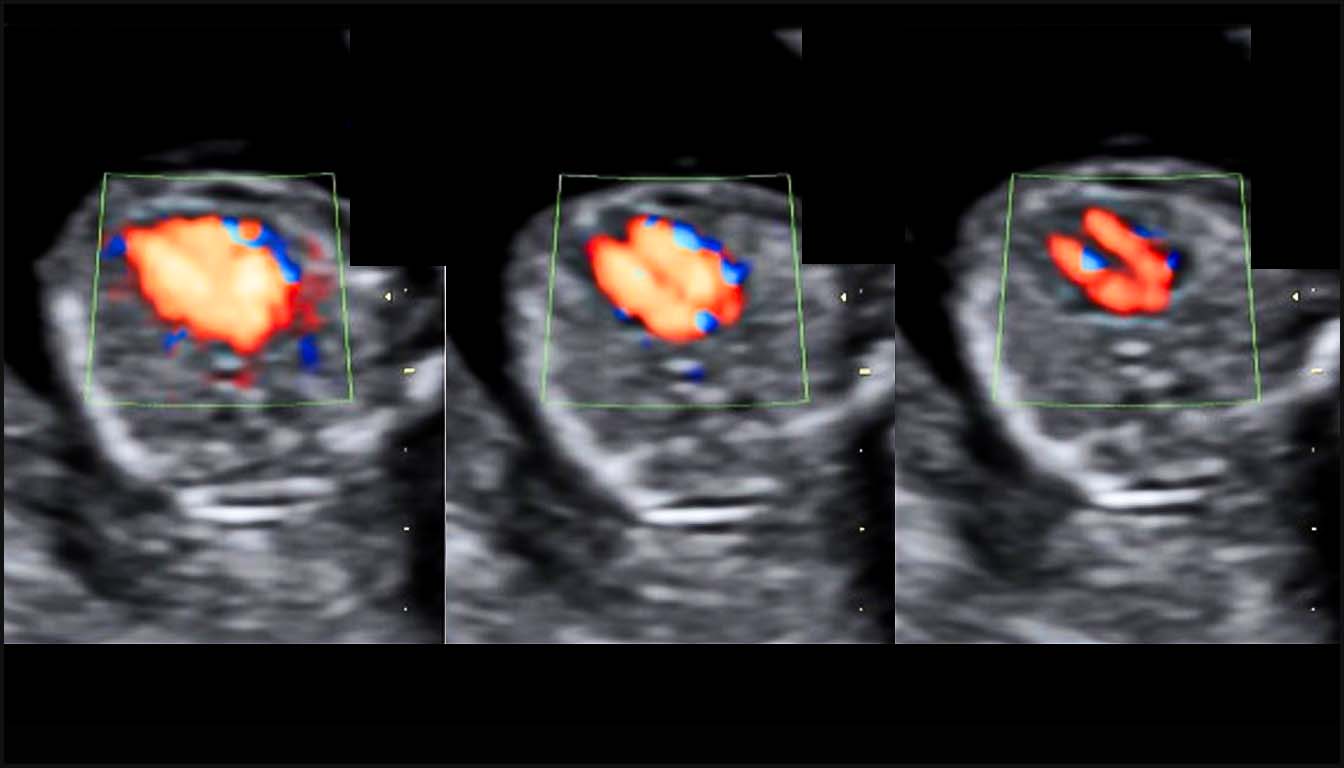
Bien que l’échocardiographie fœtale traditionnelle se déroule entre 18 et 24 semaines, la détection précoce est désormais possible grâce aux progrès de la résolution d’image et à l’utilisation systématique du Doppler couleur et pulsé. Cette approche permet de détecter plus de la moitié des anomalies cardiaques majeures dès la fin du premier trimestre, offrant ainsi un délai supplémentaire pour les tests génétiques et le conseil parental.
- Les biomarqueurs Doppler clés du premier trimestre
Le dépistage précoce repose sur trois marqueurs principaux identifiés lors de l’examen de routine : la clarté nucale (CN), le flux dans le canal d’Arantius (ductus venosus) et la recherche d’une régurgitation tricuspide.
- Le canal d’Arantius (DV) : L’évaluation par Doppler pulsé du flux veineux est un indicateur puissant de la fonction cardiaque droite. Un flux anormal, caractérisé par une onde ‘a’ inversée ou absente lors de la contraction auriculaire, est associé à un risque accru de CC structurelles, d’aneuploïdies et d’issue de grossesse défavorable. Chez les fœtus euploïdes présentant une CN augmentée, une onde ‘a’ anormale porte la prévalence des malformations cardiaques majeures à environ 15-20 %.
- La régurgitation tricuspide (TR) : La présence d’un flux rétrograde à travers la valve tricuspide pendant la systole ventriculaire est un signe d’appel majeur. Pour être cliniquement significative au premier trimestre, la TR doit être visible sur au moins la moitié de la systole avec une vélocité supérieure à 60 cm/s. La combinaison d’une CN augmentée, d’une TR et d’un flux anormal dans le DV permet d’identifier jusqu’à 55,5 % des fœtus porteurs d’une CC majeure avec un taux de faux positifs de seulement 8,8 %.
- Protocoles d’imagerie et apport du Doppler couleur
L’examen anatomique précoce s’est standardisé autour de coupes spécifiques où le Doppler couleur est considéré comme une “meilleure pratique” pour pallier la petite taille des structures cardiaques.
- La coupe des quatre cavités (4C) : Le Doppler couleur permet de visualiser deux flux d’entrée ventriculaires distincts, laminaires et de taille similaire. Il aide à diagnostiquer immédiatement des anomalies comme l’atrésie tricuspide (absence de flux à droite) ou les canaux atrioventriculaires (CAV), souvent identifiables par un flux unique en forme de “Y”.
- La coupe des trois vaisseaux et de la trachée (3VT) : Cette vue est cruciale pour évaluer les voies d’éjection et les arcs vasculaires. Normalement, l’isthme aortique et le canal artériel convergent à gauche de la trachée pour former un signe en “V” avec un flux antégrade et non turbulent. Le Doppler couleur facilite la détection précoce de la transposition des gros vaisseaux (TGV), où les vaisseaux apparaissent parallèles, et des anomalies de l’arc comme l’artère aortique droite, signalée par un signe en “U” entourant la trachée.
L’ajout du Doppler couleur à l’évaluation de la 4C et des voies d’éjection augmente significativement la sensibilité du dépistage, passant d’environ 42 % à 78 % dans les populations à bas risque.
III. Évaluation fonctionnelle et hémodynamique
Au-delà de l’anatomie, le Doppler est indispensable pour l’analyse fonctionnelle, particulièrement en cas de suspicion de dysfonction myocardique ou d’arythmie.
- Le Score de Profil Cardiovasculaire (CVP) : Ce score sur 10 points intègre des marqueurs Doppler artériels (artère ombilicale) et veineux (veine ombilicale, canal d’Arantius) pour évaluer la sévérité d’une insuffisance cardiaque. Un score CVP $\le$ 7 est prédictif d’une mortalité périnatale accrue.
- L’indice de performance myocardique (MPI) : Utilisant le Doppler pulsé pour mesurer les temps de contraction et de relaxation isovolumique, le MPI fournit une évaluation globale de la fonction ventriculaire systolique et diastolique.
- Arythmies : Le Doppler simultané de la veine cave supérieure et de l’aorte permet de documenter la relation entre les contractions auriculaires et ventriculaires, facilitant le diagnostic précis des tachycardies (ex: flutter auriculaire) ou des blocs auriculoventriculaires.
- Signes Doppler de cardiopathies critiques
Certaines anomalies “ducto-dépendantes” présentent des signes Doppler pathognomoniques essentiels pour la planification de la naissance.
- Sténose aortique critique : Elle se manifeste par un flux à haute vélocité à travers la valve aortique et, dans les cas sévères évoluant vers un syndrome d’hypoplasie du cœur gauche, par un flux rétrograde dans l’arc aortique.
- Atrésie pulmonaire : Elle est suspectée devant l’absence de flux antégrade dans l’artère pulmonaire et la présence d’un flux rétrograde dans le canal artériel, alimentant les poumons depuis l’aorte.
- Tétralogie de Fallot (TOF) : Outre la communication interventriculaire, le Doppler montre souvent une accélération du flux ou une sténose au niveau de l’artère pulmonaire, dont la taille réduite est visible dès le premier trimestre.
- Considérations techniques et sécurité
La réussite de l’examen Doppler au premier trimestre dépend de l’optimisation des réglages : une fréquence de répétition des impulsions (PRF) adaptée (19-24 cm/s), un angle d’insonation faible ($<$ 30°) et l’utilisation de sondes haute fréquence. Les opérateurs doivent respecter le principe ALARA (As Low As Reasonably Achievable), en limitant l’exposition thermique (indices thermiques et mécaniques $<$ 1.0) et la durée de l’examen Doppler à moins de 5 minutes si possible.
Conclusion
Le Doppler est l’outil indispensable qui a permis de repousser les limites du diagnostic cardiaque fœtal vers le premier trimestre. L’analyse systématique du canal d’Arantius et de la valve tricuspide, couplée à l’imagerie couleur des coupes 4C et 3VT, constitue désormais la stratégie de dépistage la plus efficace. Cette approche permet non seulement d’identifier les anomalies structurelles, mais aussi d’évaluer le retentissement fonctionnel, optimisant ainsi la prise en charge multidisciplinaire et le pronostic néonatal des enfants nés avec une cardiopathie complexe.
Doppler en échocardiographie fœtale : évaluation fonctionnelle
Dr Pierre Mace
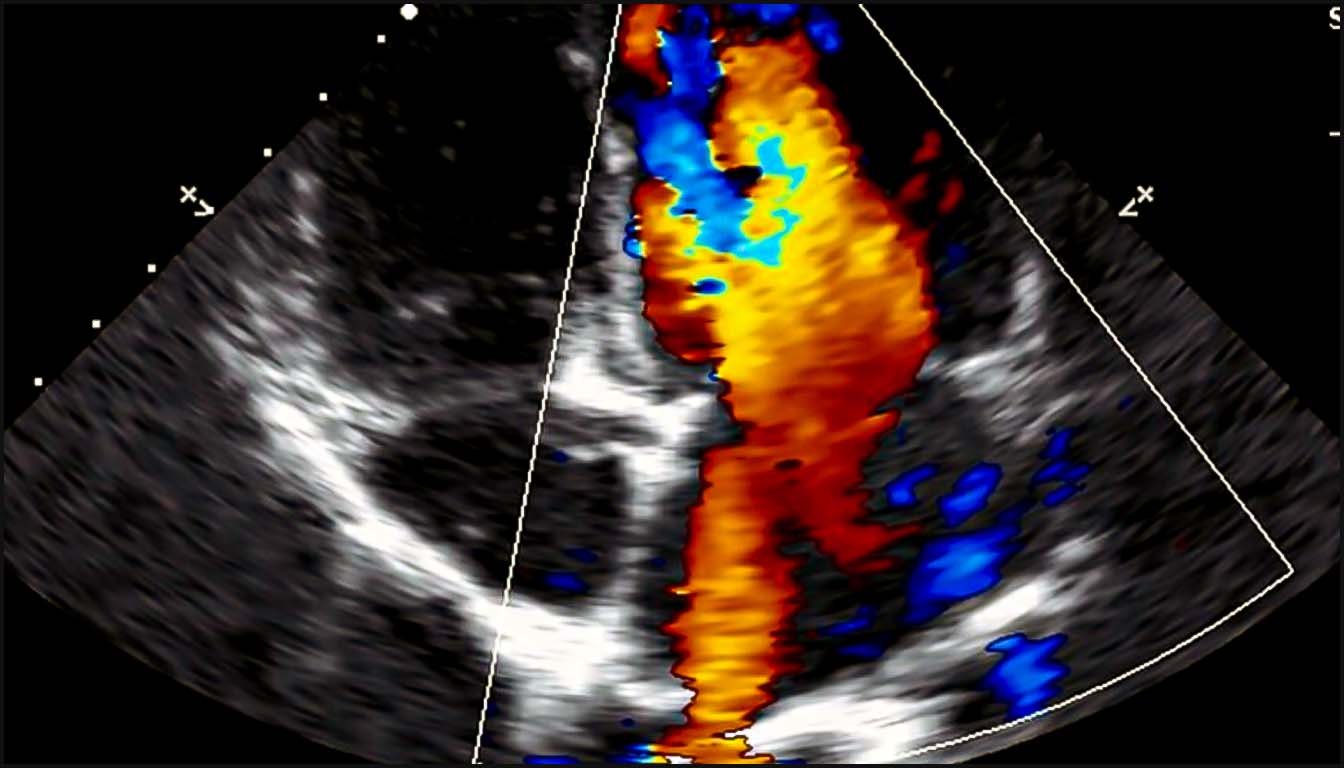
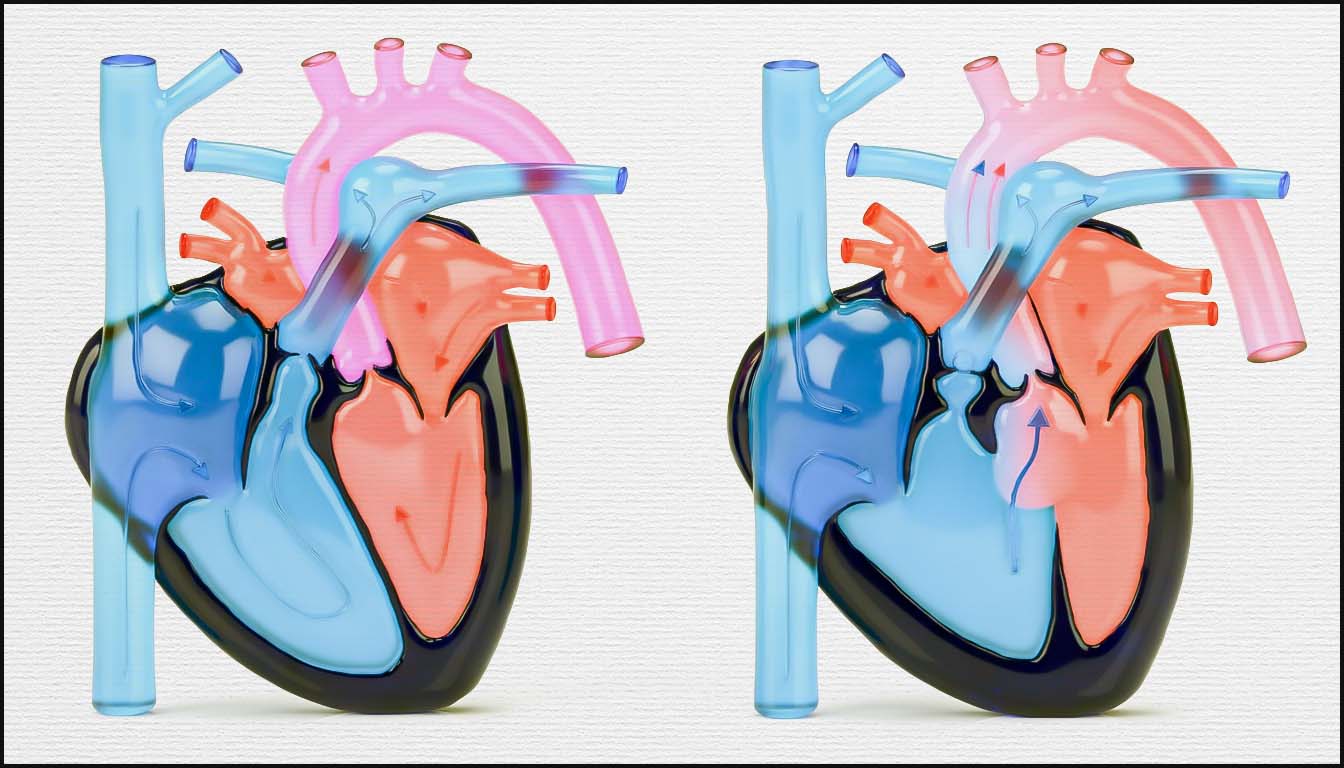
Contrairement à la circulation postnatale, le fœtus possède une circulation en parallèle où les deux ventricules travaillent ensemble pour assurer le débit cardiaque combiné, facilité par trois shunts physiologiques : le canal d’Arantius (ductus venosus), le foramen ovale et le canal artériel. Le Doppler permet de quantifier cette physiologie complexe et de détecter précocement les signes de décompensation.
- Évaluation de la fonction globale : Le score CVP et l’indice MPI
L’outil de synthèse le plus utilisé pour évaluer l’état cardiovasculaire global est le Score de Profil Cardiovasculaire (CVP). Ce score sur 10 points évalue cinq catégories majeures : l’absence d’hydrops, la taille du cœur (rapport cardiothoracique), la fonction systolique ventriculaire, et les profils Doppler veineux et artériels. Un score CVP $\le$ 7 est un indicateur de mauvais pronostic, prédisant une augmentation de la mortalité périnatale et le risque de détresse fœtale nécessitant une extraction urgente.
En complément, l’Indice de Performance Myocardique (MPI ou indice de Tei) fournit une mesure quantitative de la fonction globale, indépendante de la géométrie ventriculaire. Il est calculé en divisant la somme des temps de contraction isovolumique (ICT) et de relaxation isovolumique (IRT) par le temps d’éjection (ET). Un MPI élevé reflète une dysfonction myocardique, qu’elle soit due à une surcharge de pression (sténose), de volume (insuffisance valvulaire) ou à une cardiomyopathie primaire.
- Analyse de la fonction diastolique et pressions veineuses
La fonction diastolique fœtale est principalement évaluée par le Doppler pulsé au niveau des valves auriculo-ventriculaires (mitrale et tricuspide). Normalement, le flux présente un aspect biphasique avec une onde E (remplissage passif) et une onde A (contraction auriculaire). Une inversion du rapport E/A ou un flux monophasique sont des signes de réduction de la compliance ventriculaire ou d’augmentation des pressions de remplissage.
Le Doppler veineux est le reflet direct des pressions dans l’oreillette droite. L’analyse du canal d’Arantius (DV) est ici cruciale : un flux antégrade est normal, tandis qu’une onde ‘a’ absente ou inversée indique une augmentation de la pression veineuse centrale et une dysfonction cardiaque droite. Si la situation s’aggrave, ces anomalies de pression se transmettent à la veine ombilicale, où l’on observe alors des pulsations anormales, signe pré-terminal de défaillance cardiaque.
III. Fonction systolique et débit cardiaque
La fonction systolique est souvent évaluée de manière qualitative (contractilité globale), mais le Doppler permet des mesures précises du volume d’éjection et du débit cardiaque. Le volume d’éjection est calculé à partir du diamètre de l’anneau de la valve semi-lunaire et de l’intégrale temps-vitesse (VTI) du flux aortique ou pulmonaire. Le débit cardiaque combiné normal est d’environ 425 mL/min/kg. Des mesures sérielles sont particulièrement utiles dans les pathologies à haut débit, comme les anémies fœtales ou les malformations artérioveineuses.
L’utilisation du Doppler tissulaire (TDI) et du speckle tracking (strain) représente des techniques avancées pour analyser la mécanique myocardique de manière segmentaire. Bien que souvent réservées à la recherche, elles permettent de détecter des dysfonctions subcliniques avant que les marqueurs Doppler conventionnels ne deviennent anormaux.
- Doppler et cardiopathies ducto-dépendantes
Le Doppler joue un rôle vital dans le diagnostic des cardiopathies dites ducto-dépendantes, où la survie à la naissance dépend de la patence du canal artériel.
- Dépendance pulmonaire : En cas d’atrésie pulmonaire ou de sténose critique, le Doppler montre un flux rétrograde dans le canal artériel (de l’aorte vers l’artère pulmonaire).
- Dépendance systémique : Dans le syndrome d’hypoplasie du cœur gauche ou une coarctation sévère, on observe un flux rétrograde dans l’arc aortique et un flux de gauche à droite à travers le foramen ovale. La détection de ces flux inversés lors de l’examen fœtal est impérative pour planifier une prise en charge immédiate en salle de naissance, notamment l’administration de prostaglandines.
- Évaluation du rythme et de la conduction
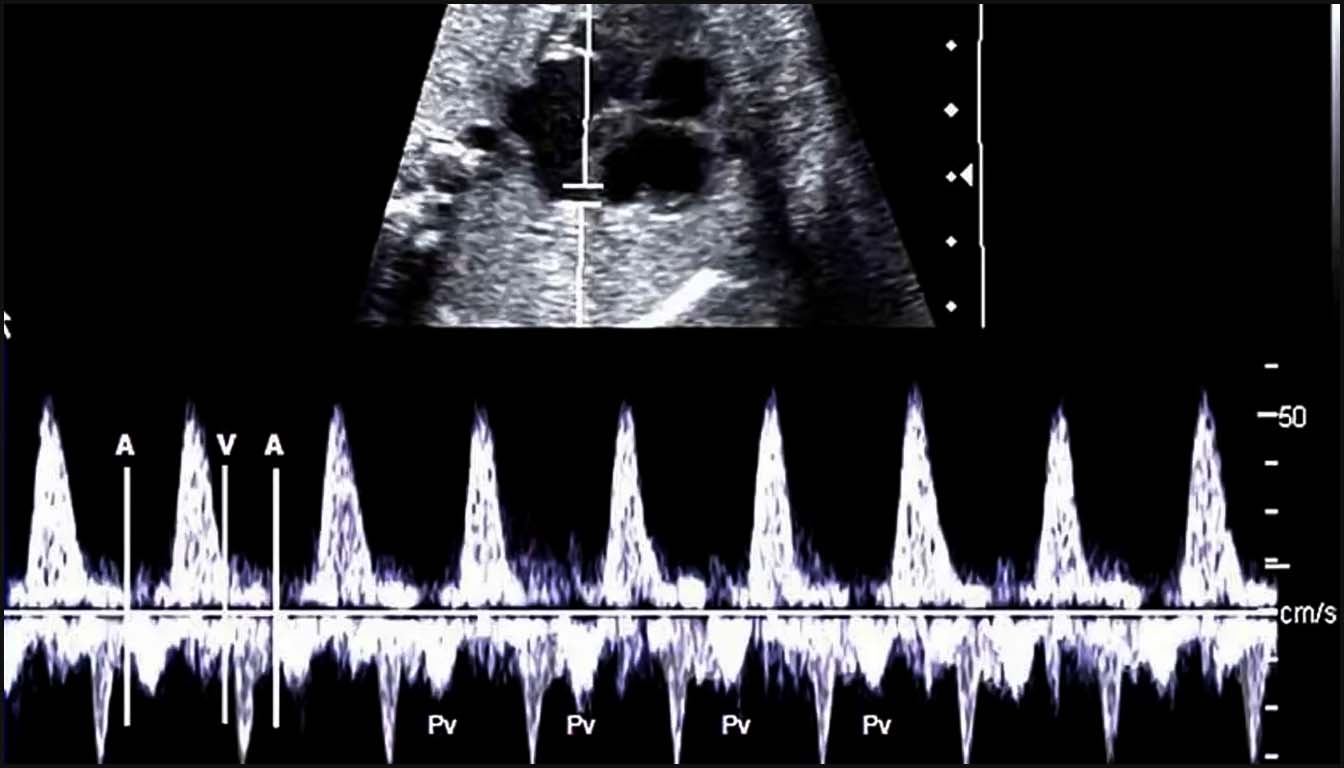
Le Doppler est l’outil de référence pour diagnostiquer les arythmies fœtales en l’absence d’ECG direct. Le Doppler pulsé simultané de la veine cave supérieure et de l’aorte permet de documenter la relation temporelle entre les événements auriculaires (onde ‘a’ de la veine) et ventriculaires (flux aortique). Cette technique permet de différencier :
- Les tachycardies supraventriculaires (SVT) par réentrée (conduction 1:1).
- Le flutter auriculaire, caractérisé par une fréquence auriculaire très élevée (jusqu’à 400 bpm) avec un bloc auriculo-ventriculaire variable.
- Le bloc auriculo-ventriculaire (BAV) complet, où l’on observe une dissociation totale entre les contractions auriculaires et ventriculaires, souvent associé à des maladies auto-immunes maternelles.
- Doppler extracardiaque et adaptation fœtale
L’évaluation fonctionnelle s’étend au-delà du cœur pour observer l’adaptation hémodynamique globale. En cas d’hypoxie ou de cardiopathie sévère, le fœtus met en place un mécanisme de “brain sparing” (épargne cérébrale). Le Doppler montre alors une diminution de la résistance dans l’artère cérébrale moyenne (ACM), signe d’une redistribution du débit cardiaque vers le cerveau au détriment des autres organes. L’analyse de l’isthme aortique par Doppler est également un indicateur sensible de l’équilibre entre les résistances vasculaires cérébrales et systémiques.
Conclusion
Le Doppler en échocardiographie fœtale ne se limite pas à valider l’anatomie ; il fournit une véritable évaluation physiologique in utero. Par la combinaison du score CVP, de l’indice MPI et de l’analyse rigoureuse des flux veineux et artériels, le clinicien peut non seulement diagnostiquer la sévérité d’une cardiopathie, mais aussi prédire le risque de défaillance cardiaque fœtale et optimiser le moment et le lieu de l’accouchement pour assurer la meilleure survie néonatale possible.
Interprétation hémodynamique : surcharge, dysfonction, redistribution
Dr PIERRE mace
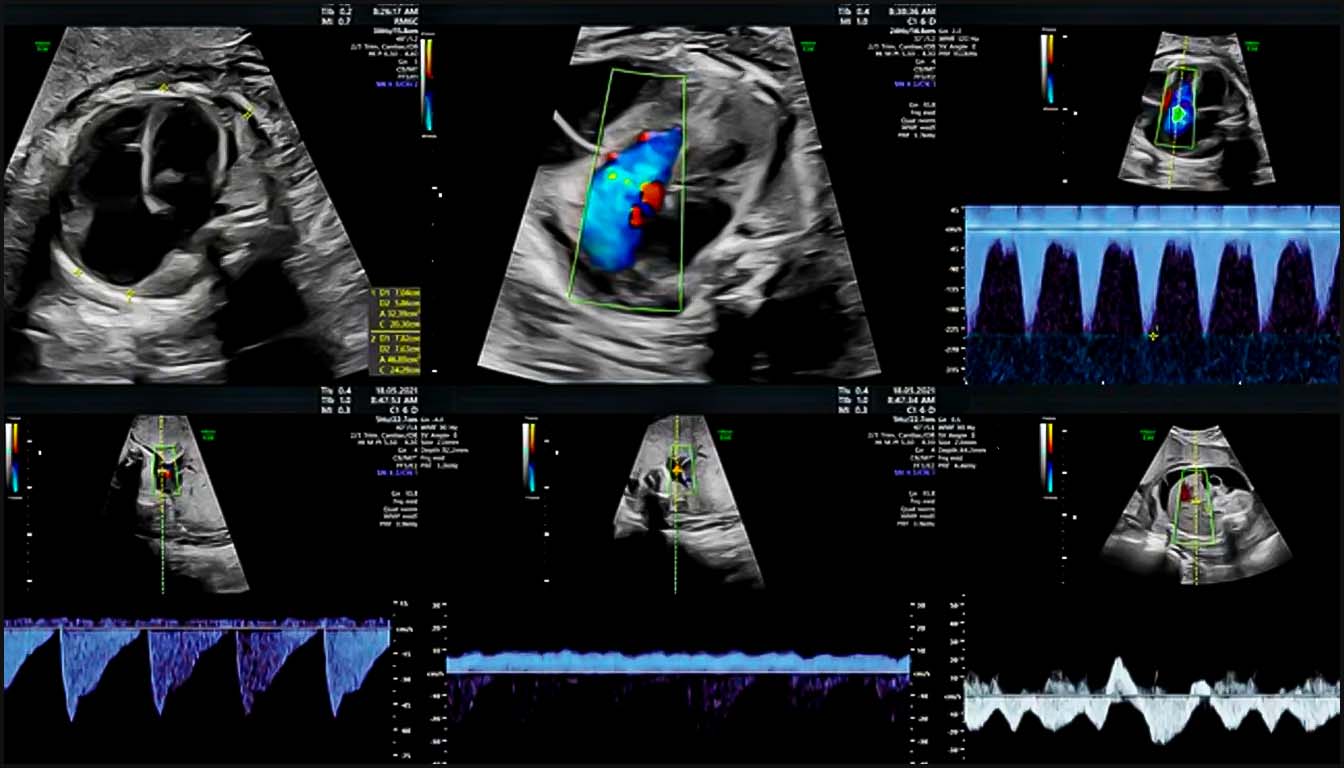
Face à une anomalie structurelle ou fonctionnelle, le fœtus met en place des mécanismes d’adaptation qui se manifestent par des phénomènes de surcharge, de dysfonction et de redistribution circulatoire.
- Les mécanismes de surcharge fœtale
La surcharge peut être de deux types : volumétrique (précharge) ou barométrique (après-charge).
- Surcharge de volume (Précharge) : Elle résulte d’une augmentation de la quantité de sang arrivant au cœur. Les causes principales incluent les insuffisances valvulaires sévères (comme dans la maladie d’Ebstein ou la dysplasie tricuspidienne), les arythmies telles que le bloc auriculo-ventriculaire complet (en raison de la bradycardie), ou encore les états d’hyperdébit. Ces états de haut débit sont souvent observés dans l’anémie fœtale, les malformations artérioveineuses, le syndrome de transfusion fœto-fœtale (STFF) ou les tumeurs vasculaires comme le tératome sacro-coccygien. La réponse initiale est une cardiomégalie (augmentation du rapport cardiothoracique), suivie potentiellement d’une défaillance cardiaque.
- Surcharge de pression (Après-charge) : Elle survient lorsqu’un ventricule doit pomper contre une résistance accrue, comme dans les sténoses valvulaires critiques (aortique ou pulmonaire) ou la coarctation de l’aorte. Ces lésions entraînent souvent une hypertrophie myocardique et peuvent évoluer vers une hypoplasie ventriculaire si le flux devient trop restreint. Une autre cause grave est la constriction du canal artériel, qui augmente brutalement la postcharge du ventricule droit.
- Évaluation et signes de dysfonction myocardique
L’identification d’une dysfonction est cruciale pour le pronostic. Elle est évaluée par une combinaison d’imagerie structurelle et de Doppler.
- Le Score de Profil Cardiovasculaire (CVP) : C’est l’outil de référence pour quantifier la sévérité de l’atteinte hémodynamique. Ce score sur 10 points évalue l’hydrops, la taille du cœur, la fonction ventriculaire, et les Dopplers artériels et veineux. Un score CVP $\le$ 7 est un indicateur fort de risque accru de mortalité périnatale et de détresse in utero.
- Marqueurs Doppler de dysfonction :
- Dysfonction systolique : Elle se manifeste par une diminution de la contractilité ventriculaire ou par l’apparition de régurgitations auriculo-ventriculaires (tricuspide ou mitrale). L’indice de performance myocardique (MPI) ou indice de Tei permet une évaluation globale de la fonction systolo-diastolique.
- Dysfonction diastolique : Elle précède souvent la défaillance systolique et se traduit par des profils de remplissage ventriculaire anormaux (flux monophasique aux valves AV).
- Signes veineux terminaux : L’inversion de l’onde ‘a’ dans le canal d’Arantius (DV) et l’apparition de pulsations dans la veine ombilicale sont des signes d’une augmentation critique de la pression auriculaire droite et de l’insuffisance cardiaque terminale.
III. Redistribution circulatoire et “Brain Sparing”
Lorsque le fœtus est confronté à une hypoxie chronique ou à une cardiopathie compromettant le débit systémique, il active des mécanismes de redistribution pour protéger les organes nobles, principalement le cerveau.
- Le phénomène de “Brain Sparing” : Ce mécanisme se manifeste par une vasodilatation des artères cérébrales pour augmenter l’apport en oxygène. À l’échographie Doppler, on observe une diminution de l’indice de pulsatilité (PI) de l’artère cérébrale moyenne (ACM). Un rapport artère ombilicale / ACM > 1 est un signe d’alerte important de redistribution.
- Isthme aortique : Il joue le rôle de “pont” entre les deux circulations ventriculaires. Un flux diastolique rétrograde dans l’isthme aortique identifie les fœtus avec une redistribution cérébrale significative.
- Impact sur le neurodéveloppement : Des études récentes par IRM fœtale montrent que les perturbations de l’hémodynamique cérébrale, notamment une diminution du flux dans la veine cave supérieure (SVC) ou une réduction de la délivrance d’oxygène au cerveau (CDO2), sont directement associées à un risque accru de retard neurodéveloppemental postnatale, indépendamment de la chirurgie.
- Conséquences cliniques et prise en charge
L’interprétation correcte de ces signes hémodynamiques guide toute la stratégie périnatale :
- Surveillance sérielle : Elle est impérative dans les lésions obstructives pour détecter une progression vers une dysfonction irréversible ou un hydrops.
- Interventions in utero : Dans certains cas de sténose aortique critique avec flux rétrograde dans l’arche, une valvuloplastie fœtale peut être discutée pour prévenir l’évolution vers un syndrome d’hypoplasie du cœur gauche et tenter de restaurer une hémodynamique biventriculaire.
- Planification de la naissance : La présence d’une dysfonction sévère ou d’un hydrops impose un accouchement dans un centre de niveau 3 ou 4 avec une équipe de réanimation cardiaque prête à intervenir immédiatement (septostomie, prostaglandines).
Conclusion
L’interprétation hémodynamique chez le fœtus dépasse le simple diagnostic anatomique. Elle nécessite une analyse dynamique de la surcharge ventriculaire, le monitorage rigoureux de la dysfonction myocardique via le score CVP, et la reconnaissance précoce de la redistribution cérébrale. Cette approche systématique est le seul moyen d’optimiser le devenir vital et neurodéveloppemental des enfants nés avec une cardiopathie congénitale complexe.
Cardiopathies congénitales fréquentes :
stratégie diagnostique
Dr Bertrand Stos
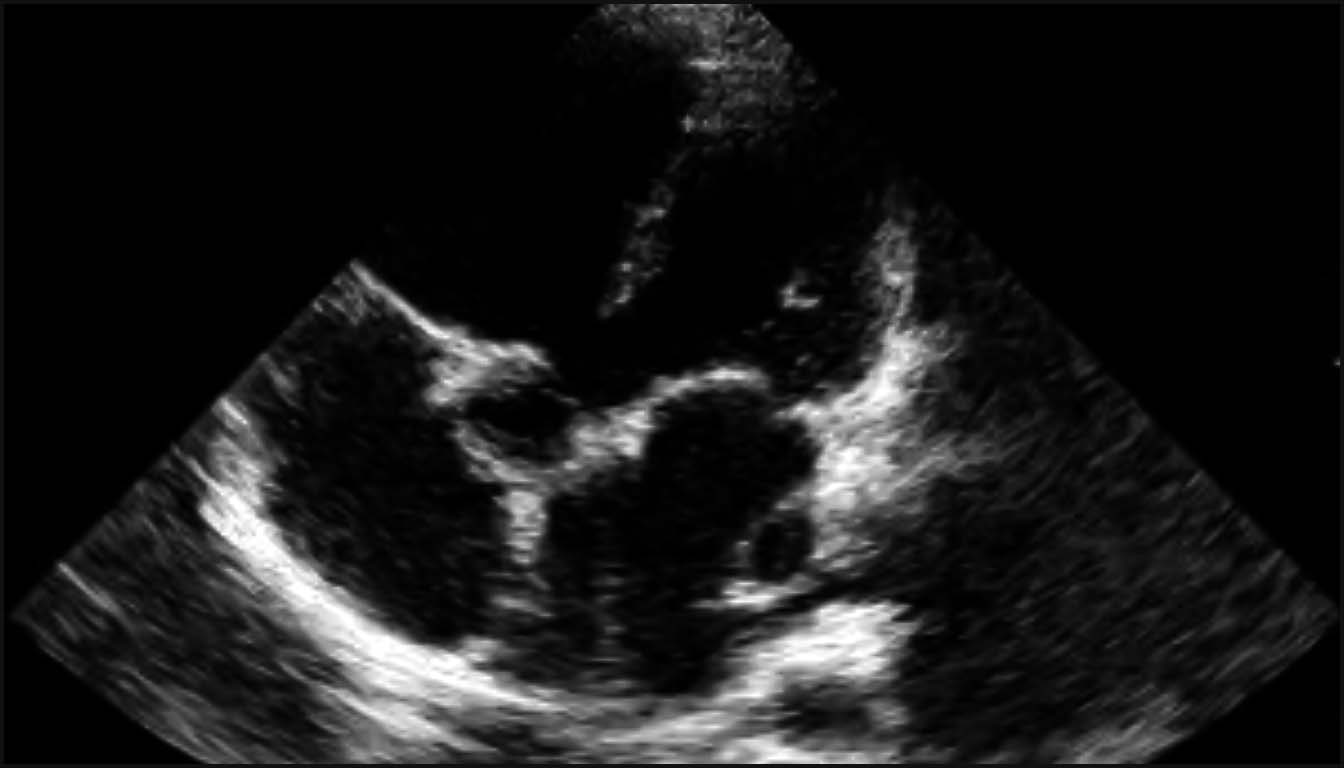
Malgré les progrès de l’imagerie, le diagnostic prénatal reste un défi, car environ 90 % des cas surviennent dans la population à bas risque, sans facteurs prédisposants identifiables. Une stratégie diagnostique rigoureuse, allant du dépistage de routine à l’échocardiographie spécialisée, est donc indispensable pour optimiser la prise en charge néonatale.
- Évolution de la stratégie de dépistage de routine
Le dépistage s’effectue traditionnellement lors de l’échographie morphologique du deuxième trimestre (18-22 semaines). Historiquement, l’examen se limitait à la coupe des quatre cavités (4C), mais celle-ci ne permet de détecter que 26 % à 50 % des malformations cardiaques majeures, manquant notamment les anomalies des gros vaisseaux.
La recommandation actuelle est de passer à une approche de cinq coupes axiales essentielles :
- Le situs abdominal : vérification de la position de l’estomac et des vaisseaux abdominaux.
- La coupe des quatre cavités : évaluation de la taille, de l’axe cardiaque (45° ± 20°) et de la symétrie des ventricules et des oreillettes.
- Les voies d’éjection gauche et droite (LVOT/RVOT) : indispensables pour détecter les anomalies conotroncales.
- La coupe des trois vaisseaux et de la trachée (3VT) : elle permet de visualiser l’artère pulmonaire, l’aorte et la veine cave supérieure, formant normalement un signe en “V” à gauche de la trachée.
L’intégration systématique des voies d’éjection et de la coupe 3VT permet d’augmenter le taux de détection global jusqu’à 90 %.
- Stratégie de détection précoce au premier trimestre
Les avancées technologiques permettent désormais d’identifier plus de la moitié des CC majeures dès 11 à 13 semaines de gestation. Cette détection précoce repose sur l’utilisation de marqueurs spécifiques :
- La clarté nucale (CN) : une CN augmentée ($\ge$ 3,5 mm) est un signe d’appel majeur, même avec un caryotype normal.
- Le Doppler du canal d’Arantius (ductus venosus) : une onde ‘a’ inversée ou absente signale une potentielle dysfonction cardiaque.
- La régurgitation tricuspide : sa présence au premier trimestre multiplie significativement le risque de cardiopathie structurelle.
L’utilisation combinée de ces marqueurs et du Doppler couleur sur les coupes 4C et 3VT permet de suspecter des pathologies complexes dès la fin du premier trimestre, offrant ainsi plus de temps pour le conseil génétique.
III. Échocardiographie fœtale spécialisée et évaluation fonctionnelle
Lorsqu’une anomalie est suspectée, une échocardiographie détaillée doit être réalisée par un expert. Elle repose sur une analyse segmentaire séquentielle qui définit précisément l’anatomie : connexions veineuses, types de valves et relations entre les ventricules et les gros vaisseaux.
Au-delà de l’anatomie, l’évaluation de la fonction cardiaque est cruciale pour le pronostic. Le Score de Profil Cardiovasculaire (CVP) est l’outil de référence. Noté sur 10 points, il évalue cinq signes de décompensation :
- Présence d’hydrops (épanchements).
- Cardiomégalie (rapport cardiothoracique).
- Fonction systolique ventriculaire.
- Profil Doppler artériel (artère ombilicale).
- Profil Doppler veineux (veine ombilicale et canal d’Arantius).
Un score CVP $\le$ 7 est prédictif d’une mortalité périnatale accrue et d’un risque de détresse in utero.
- Signes diagnostiques des cardiopathies les plus fréquentes
Les sources permettent de dégager des schémas types pour les CC majeures :
- Tétralogie de Fallot (TOF) : Elle se caractérise par une communication interventriculaire (CIV) large, une aorte “à cheval” sur le septum et une artère pulmonaire de petit calibre.
- Transposition des gros vaisseaux (TGA) : Le signe pathognomonique est l’émergence des vaisseaux en parallèle plutôt que leur croisement normal. Au Doppler, une seule artère est souvent visible dans le plan 3VT.
- Syndrome d’hypoplasie du cœur gauche (HLHS) : On observe un ventricule gauche minuscule ou invisible, une atrésie mitrale/aortique et, signe Doppler clé, un flux rétrograde dans l’arc aortique sur la coupe 3VT.
- Canal atrioventriculaire (CAV) : Typiquement associé à la trisomie 21, il présente une valve unique commune, un défaut septal central et un flux Doppler d’entrée ventriculaire en forme de “Y”.
- Intégration génétique et extracardiaque
Le diagnostic d’une CC doit systématiquement déclencher la recherche d’anomalies associées. Environ 15 % à 30 % des fœtus porteurs de CC présentent une anomalie chromosomique. Les conotroncales (TOF, tronc artériel commun) sont particulièrement liées à la microdélétion 22q11.2 (syndrome de DiGeorge). L’évaluation doit donc inclure un conseil génétique et, selon les cas, un caryotype, une analyse par puce à ADN (CMA) ou un séquençage de l’exome.
Conclusion
La stratégie diagnostique actuelle repose sur une surveillance continue tout au long de la grossesse. L’approche systématique des cinq coupes axiales en dépistage de routine, renforcée par les biomarqueurs du premier trimestre et une évaluation fonctionnelle par le score CVP, transforme le pronostic fœtal. Cette rigueur permet de planifier l’accouchement dans des centres spécialisés, évitant ainsi une décompensation hémodynamique fatale à la naissance pour les nouveau-nés porteurs de cardiopathies ducto-dépendantes.
Cardiopathies congénitales complexes :
approche simplifiée
Dr Bertrand Stos
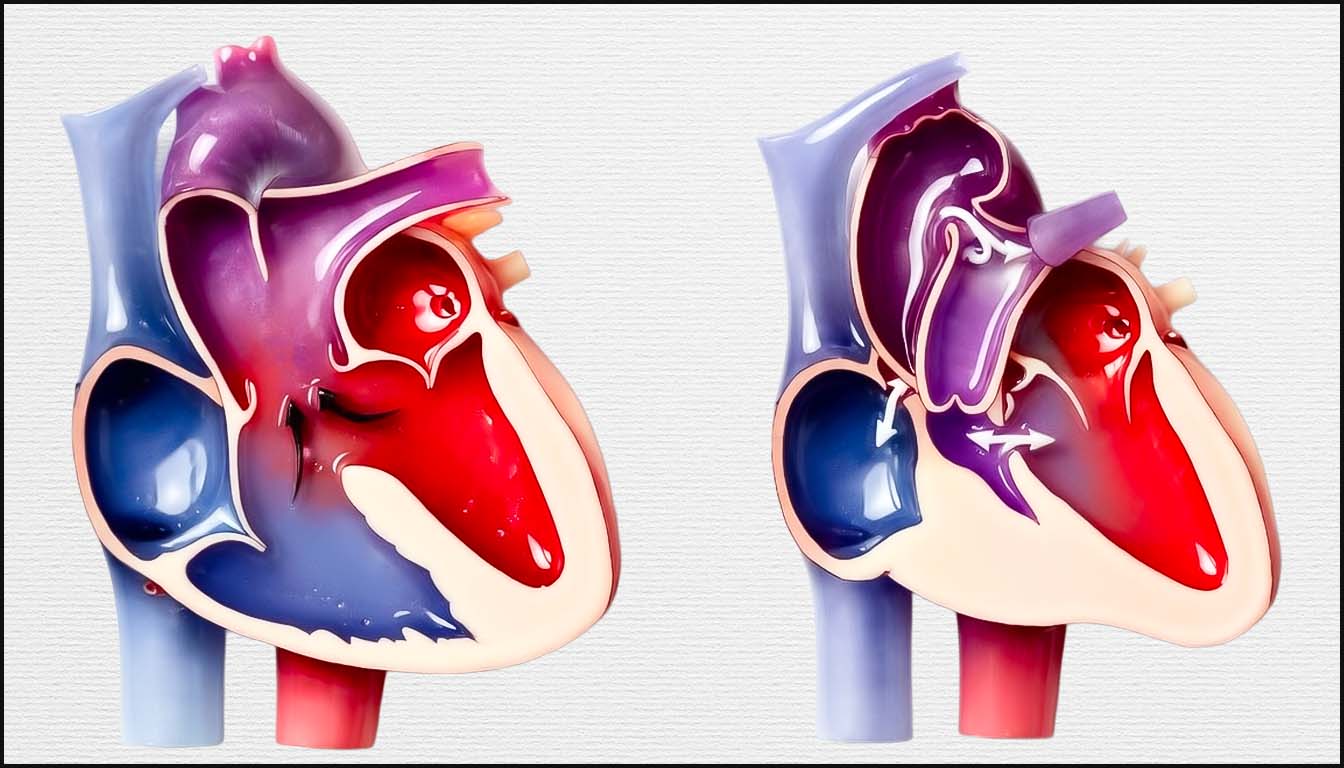
Une approche simplifiée et systématique, reposant sur des protocoles d’imagerie standardisés et une analyse segmentaire rigoureuse, est essentielle pour améliorer les taux de détection prénatale.
- Le protocole des cinq coupes axiales : une base universelle
L’approche simplifiée la plus efficace repose sur la réalisation de cinq coupes axiales transversales essentielles, allant de l’abdomen supérieur jusqu’à l’entrée du thorax. Cette méthode, recommandée pour le dépistage de routine, inclut : le situs abdominal, la coupe des quatre cavités (4C), les voies d’éjection ventriculaires gauche et droite, et enfin la coupe des trois vaisseaux et de la trachée (3VT).
Historiquement, l’examen se limitait à la coupe 4C, mais celle-ci ne permet de détecter qu’environ 50 % des anomalies majeures, manquant fréquemment les anomalies des gros vaisseaux comme la transposition des gros vaisseaux (TGA) ou la coarctation de l’aorte. L’intégration systématique des voies d’éjection et de la coupe 3VT permet de porter le taux de détection à près de 90 % dans les centres expérimentés. La coupe 3VT est particulièrement utile pour comparer la taille relative de l’aorte et de l’artère pulmonaire, et pour vérifier que les deux arcs convergent normalement en forme de « V » à gauche de la trachée.
- Dépistage précoce et marqueurs du premier trimestre
Les avancées technologiques permettent désormais de suspecter des cardiopathies complexes dès 11 à 13 semaines de gestation. Une approche simplifiée au premier trimestre repose sur la combinaison de la mesure de la clarté nucale (CN) et de l’évaluation Doppler du flux dans le canal d’Arantius (ductus venosus) et à travers la valve tricuspide.
Une CN augmentée ($\ge$ 3,5 mm) ou un flux inversé de l’onde « a » dans le canal d’Arantius sont des signes d’alerte majeurs qui imposent une échocardiographie fœtale détaillée. De plus, la présence d’une régurgitation tricuspide significative à ce stade augmente considérablement la probabilité d’une anomalie cardiaque structurelle majeure. L’utilisation du Doppler couleur sur les coupes 4C et 3VT dès le premier trimestre permet d’identifier plus de 50 % des malformations cardiaques majeures avec un taux de faux positifs inférieur à 9 %.
III. L’analyse segmentaire simplifiée : une logique de connexion
Pour simplifier le diagnostic des lésions complexes, l’examen doit suivre une analyse segmentaire séquentielle. Cette méthode consiste à définir trois segments : le situs auriculaire, les connections atrio-ventriculaires (AV) et les connections ventriculo-artérielles (VA).
- Le Situs : On vérifie d’abord que l’estomac et le cœur sont bien situés à gauche. Un axe cardiaque anormal (normal : 45° $\pm$ 20°) est souvent le premier signe d’une cardiopathie complexe.
- Connections AV : La coupe 4C permet d’évaluer la symétrie des ventricules et l’intégrité de la croix du cœur. Un déséquilibre de taille entre le ventricule gauche (VG) et le ventricule droit (VD) doit faire suspecter un syndrome d’hypoplasie du cœur gauche (HLHS) ou une coarctation de l’aorte.
- Connections VA : On vérifie que les gros vaisseaux se croisent normalement à leur origine. L’absence de croisement (vaisseaux parallèles) est le signe caractéristique de la TGA.
- Reconnaissance par « patterns » de flux Doppler
Une approche diagnostique innovante consiste à identifier des signatures visuelles ou « patterns » répétitifs au Doppler couleur sur les coupes 4C et 3VT.
- Pattern HLHS : Sur la coupe 4C, on n’observe qu’un seul flux ventriculaire entrant (le droit), tandis qu’en 3VT, on visualise un vaisseau unique avec un flux rétrograde (bleu) dans l’arc aortique extrêmement hypoplasique.
- Pattern TGA : Les deux vaisseaux d’éjection émergent parallèlement du cœur ; en 3VT, on ne voit souvent qu’un seul vaisseau central avec un trajet long et rectiligne vers le médiastin.
- Pattern Fallot / DORV : On observe un large défaut septal avec une aorte « à cheval » sur le septum interventriculaire ; en 3VT, le vaisseau pulmonaire est typiquement plus étroit que l’aorte.
- Pattern AVSD : Un flux d’entrée unique en forme de « Y » remplit les deux ventricules simultanément, traduisant la présence d’une valve AV commune.
- Stratification du risque et gestion périnatale
Une fois la cardiopathie complexe identifiée, l’approche simplifiée se déplace vers la planification de la naissance en fonction de la stabilité hémodynamique attendue. Le système de stratification par niveaux de soins (LOC) guide cette décision :
- LOC 1 et 2 : Cardiopathies sans risque immédiat d’instabilité, permettant une naissance dans un hôpital local avec accès à une consultation cardiologique.
- LOC 3 et 4 : Cardiopathies critiques (ex: TGA avec septum intact, HLHS avec foramen ovale restrictif) nécessitant une intervention immédiate en salle de naissance ou par cathétérisme urgent.
Pour les lésions ducto-dépendantes, comme l’atrésie pulmonaire ou la coarctation sévère, l’initiation précoce de prostaglandines (PGE) est cruciale pour maintenir la circulation systémique ou pulmonaire jusqu’à la chirurgie. En cas d’arythmie complexe ou de bloc auriculo-ventriculaire complet, des outils avancés comme la magnétocardiographie fœtale (fMCG) peuvent affiner le diagnostic et guider le traitement médical in utero.
Conclusion
L’adoption d’une approche systématique simplifiée, passant par le protocole des cinq coupes axiales et la reconnaissance de « patterns » Doppler spécifiques, permet de transformer radicalement le pronostic des cardiopathies congénitales complexes. Cette stratégie permet non seulement un diagnostic anatomique plus fiable, mais elle assure surtout une transition sécurisée vers la vie postnatale grâce à une stratification précise du risque et une prise en charge multidisciplinaire coordonnée dès la salle de naissance.
Corrélation structure – hémodynamique – pronostic
Pr Yves Ville
La corrélation entre la structure cardiaque, le profil circulatoire et le pronostic vital et neurodéveloppemental est désormais le pilier de la cardiologie fœtale moderne.
- De la structure à l’hémodynamique : la cascade des anomalies
Le diagnostic commence par une analyse segmentaire rigoureuse, incluant la coupe des quatre cavités (4C), les voies d’éjection et la coupe des trois vaisseaux et de la trachée (3VT). Une anomalie structurelle initiale déclenche souvent des modifications hémodynamiques progressives. Par exemple, une sténose aortique (SA) critique peut restreindre le flux ventriculaire gauche, entraînant une fibroélastose endocardique et évoluant in utero vers un syndrome d’hypoplasie du cœur gauche (SHCG).
La détection de flux anormaux par Doppler couleur est un marqueur structurel et fonctionnel essentiel. Un flux rétrograde dans l’isthme aortique ou le canal artériel est le signe pathognomonique d’une pathologie ducto-dépendante, indiquant que la survie néonatale dépendra de la patence du canal artériel. À l’inverse, l’asymétrie des cavités (VD > VG) peut être le seul signe d’appel d’une coarctation de l’aorte, bien que sa valeur prédictive isolée reste modérée.
- L’évaluation hémodynamique globale : le score CVP
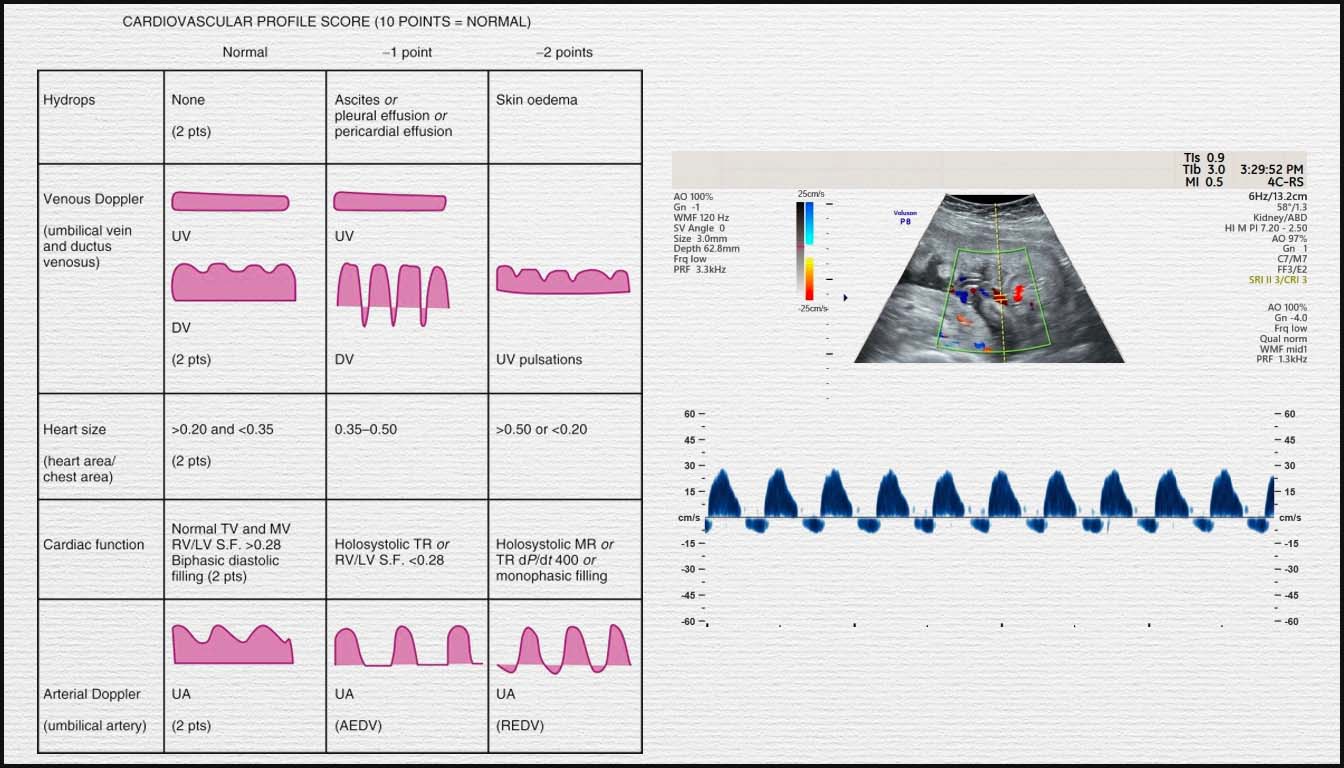
Le pronostic fœtal ne dépend pas uniquement de la lésion anatomique, mais de la capacité du cœur à maintenir un débit cardiaque suffisant. Le Score de Profil Cardiovasculaire (CVP) synthétise cette corrélation sur 10 points en évaluant l’hydrops, la taille du cœur (rapport cardiothoracique), la fonction ventriculaire et les flux Doppler artériels et veineux.
Une corrélation directe existe entre un score CVP $\le$ 7 et une augmentation significative de la mortalité périnatale. Les signes de décompensation les plus graves incluent l’inversion de l’onde ‘a’ dans le canal d’Arantius (ductus venosus) et l’apparition de pulsations dans la veine ombilicale, reflets d’une hypertension auriculaire droite et d’une insuffisance cardiaque terminale. De même, une régurgitation tricuspide (RT) holosystolique précoce est un marqueur fort de malformation structurelle majeure et d’aneuploïdie.
III. L’axe cœur-cerveau : hémodynamique et devenir neurologique
L’une des avancées les plus marquantes est la corrélation entre les perturbations hémodynamiques fœtales et le développement cérébral. Les fœtus porteurs de CC complexes, comme la transposition des gros vaisseaux (TGA) ou le SHCG, présentent des altérations de la perfusion cérébrale détectables in utero.
Le mécanisme de “brain sparing” (épargne cérébrale), caractérisé par une vasodilatation de l’artère cérébrale moyenne (ACM) et une baisse de son indice de pulsatilité (PI), est une réponse compensatoire à l’hypoxie. Cependant, des études récentes par IRM fœtale montrent que les fœtus avec le débit le plus réduit dans la veine cave supérieure (SVC), reflet du débit cérébral, ont les scores cognitifs et moteurs les plus bas à 18 mois. De plus, des anomalies des métabolites cérébraux (rapport NAA/Choline abaissé) émergent au troisième trimestre, période de croissance cérébrale exponentielle, suggérant que le substrat métabolique est insuffisant pour soutenir une maturation corticale normale.
- Stratification du risque et planification du pronostic
La corrélation structure-hémodynamique permet de classer les fœtus par Niveaux de Soins (LOC), optimisant ainsi la survie néonatale.
- LOC 2 : Lésions ducto-dépendantes stables (SA sévère, coarctation) nécessitant des prostaglandines à la naissance.
- LOC 4 : Cardiopathies critiques avec instabilité immédiate (TGA avec septum intact, SHCG avec septum interauriculaire restrictif) imposant une naissance dans un centre de chirurgie cardiaque avec intervention immédiate (septostomie atriale).
Dans le cas des cœurs univentriculaires, certains facteurs structurels comme la dominance du ventricule droit, la présence d’hydrops ou d’anomalies extracardiaques sont associés à une survie sans transplantation significativement plus faible (72 % à 4 ans pour la cohorte globale). Pour la SA critique, des modèles prédictifs basés sur le rapport de longueur RV/LV et la vélocité de la régurgitation mitrale (MR-Vmax) permettent désormais de sélectionner les candidats idéaux pour une valvuloplastie aortique fœtale (VAF), avec une probabilité de survie biventriculaire (BV) atteignant 96 % dans les cas favorables.
Conclusion
La synthèse entre l’imagerie structurelle et les mesures hémodynamiques dynamiques transforme le conseil prénatal. Le pronostic d’un fœtus porteur d’une CC ne se définit plus uniquement par son étiquette diagnostique, mais par son profil circulatoire global et son impact sur la croissance des organes, particulièrement le cerveau. Cette approche multidisciplinaire, intégrant l’échocardiographie, l’IRM et le score CVP, permet une prise en charge “sur mesure” qui améliore non seulement la survie immédiate mais aussi la qualité de vie à long terme des enfants nés avec une cardiopathie complexe.
Troubles du rythme et de la conduction fœtale
Dr Bertrand Stos
Cette synthèse détaille l’approche systématique du diagnostic, de l’évaluation pronostique et de la prise en charge de ces pathologies.
- Arsenal diagnostique : de l’échographie à la magnétocardiographie
Le diagnostic repose initialement sur l’échographie fœtale, qui utilise le mode M et le Doppler pulsé pour analyser la relation temporelle entre les contractions auriculaires et ventriculaires. Les techniques de Doppler simultané (Veine cave supérieure/Aorte ou Influx mitral/Efflux aortique) sont indispensables pour définir le mécanisme de l’arythmie, en mesurant l’intervalle mécanique PR (conduction AV).
Une avancée technologique majeure est la magnétocardiographie fœtale (fMCG). Bien que sa disponibilité soit limitée, elle permet un enregistrement continu (type Holter) du signal électromagnétique du cœur fœtal. La fMCG est supérieure à l’échographie pour détecter les anomalies de repolarisation, comme le syndrome du QT long (SQTL), et pour identifier précisément les torsades de pointes (TdP), souvent confondues avec d’autres rythmes complexes à l’échographie.
- Bradyarythmies et troubles de la conduction
La bradycardie fœtale est définie par une fréquence cardiaque soutenue inférieure à 110-120 bpm.
- Le Bloc Auriculo-Ventriculaire (BAV) complet : C’est la forme la plus sévère, touchant 1 grossesse sur 14 000 à 20 000. Deux étiologies prédominent :
- L’origine immunologique (40 %) : Liée au passage transplacentaire d’anticorps maternels anti-Ro/SSA et anti-La/SSB qui attaquent le nœud AV et les fibres de Purkinje. Elle présente un risque de cardiomyopathie associée.
- L’origine structurelle (50-55 %) : Souvent associée à des malformations complexes comme l’hétérotaxie (isomérisme gauche) ou le canal atrioventriculaire.
- Pronostic et gestion : Le pronostic est particulièrement réservé en cas de cardiopathie structurelle ou si la fréquence ventriculaire est inférieure à 55 bpm. Le traitement par dexaméthasone peut être tenté pour réduire l’inflammation dans les blocs de 1er ou 2e degré, tandis que les bêta-mimétiques (terbutaline) sont utilisés pour augmenter la fréquence ventriculaire.
III. Tachyarythmies fœtales
Une fréquence cardiaque supérieure à 160-180 bpm définit la tachycardie. Elle peut mener rapidement à un hydrops fœtal (épanchements, œdème) par insuffisance cardiaque congestive.
- Tachycardie Supraventriculaire (SVT) : C’est l’arythmie sévère la plus fréquente, souvent due à une réentrée auriculoventriculaire (AVRT). La fréquence se situe généralement entre 220 et 250 bpm.
- Flutter Auriculaire : Caractérisé par une fréquence auriculaire extrêmement rapide (jusqu’à 400 bpm) avec un aspect en “dents de scie” au mode M et un degré variable de bloc de conduction (souvent 2:1).
- Tachycardie Ventriculaire (TV) : Plus rare et grave, elle est souvent associée à un SQTL ou à une myocardite.
- Stratégies de traitement transplacentaire
L’objectif du traitement est de restaurer un rythme sinusal ou, à défaut, une fréquence ventriculaire compatible avec une hémodynamique normale pour résoudre l’hydrops.
- Choix médicamenteux :
- Pour la SVT, le Flécaïnide est de plus en plus privilégié comme traitement de première intention (efficacité de 90-96 % sans hydrops), suivi par la Digoxine et le Sotalol.
- Pour le Flutter, le Sotalol ou la Digoxine sont recommandés en première ligne.
- En cas d’hydrops, l’absorption intestinale maternelle et le transfert placentaire sont diminués, nécessitant souvent des combinaisons thérapeutiques (ex: Digoxine + Flécaïnide) ou, plus rarement, des injections fœtales directes.
- Sécurité : Un monitorage maternel (ECG, taux sériques) est impératif pour éviter la toxicité médicamenteuse, particulièrement avec le Flécaïnide et le Sotalol qui peuvent allonger l’intervalle QT maternel.
- Évaluation fonctionnelle et Score CVP
Le Score de Profil Cardiovasculaire (CVP) est l’outil pronostique de référence. Il évalue sur 10 points cinq catégories : hydrops, taille du cœur, fonction ventriculaire, et flux Doppler artériel et veineux. Un score CVP $\le$ 7 indique un risque élevé de mortalité périnatale et de détresse in utero, guidant ainsi la décision d’extraction fœtale si l’âge gestationnel le permet.
- Planification de la naissance et devenir
La prise en charge doit être coordonnée par une équipe multidisciplinaire (“Cardio-obstetrics team”).
- Niveaux de soins (LOC) : Les arythmies instables ou compliquées d’hydrops relèvent du niveau 4 (LOC 4), imposant un accouchement dans un centre de chirurgie cardiaque avec des spécialistes (néonatologistes, cardiologues pédiatres) présents en salle de naissance.
- Interventions immédiates : Pour les BAV sévères ou les tachycardies non contrôlées, des mesures d’urgence comme l’administration d’agents chronotropes, une cardioversion immédiate ou la mise en place d’un stimulateur cardiaque néonatal (pacing) doivent être prévues.
Enfin, l’impact des troubles du rythme prolongés sur le développement cérébral est une préoccupation émergente. Des études montrent qu’une réduction du débit dans la veine cave supérieure (SVC), reflet du flux cérébral, est corrélée à des scores neurodéveloppementaux plus faibles à 18 mois, soulignant l’importance d’un contrôle rapide de l’arythmie in utero.
Conclusion
La gestion des arythmies fœtales a bénéficié de l’amélioration des protocoles de traitement transplacentaire et de l’apport diagnostique de la fMCG. Une approche systématique intégrant une analyse précise du rythme, un monitorage rigoureux par le score CVP et une planification de naissance hautement spécialisée permet aujourd’hui d’atteindre une survie globale élevée pour ces patients fragiles.
Évaluation pronostique en cardiologie fœtale
Pr Yves Ville
Cette synthèse détaille les outils et les marqueurs identifiés dans les sources pour établir une stratégie pronostique rigoureuse.
- Marqueurs de dépistage précoce et risque de cardiopathie majeure
Dès le premier trimestre (11-13 semaines), certains signes échographiques constituent les premiers indicateurs pronostiques. La clarté nucale (CN) est le marqueur le plus robuste : un seuil au-delà du 95e percentile multiplie le risque de cardiopathie congénitale (CC). Le risque de malformation cardiaque majeure atteint environ 15 % lorsque la CN est ≥ 3,5 mm et que le flux dans le canal d’Arantius (ductus venosus) est anormal, et grimpe à 20 % si la CN dépasse le 99e percentile.
La présence d’une régurgitation tricuspide (RT) à ce stade est également un signe d’alerte majeur. La combinaison d’une CN augmentée, d’une RT et d’un flux anormal dans le canal d’Arantius permet de détecter plus de la moitié des CC majeures. Ces marqueurs précoces permettent d’orienter rapidement les fœtus vers une échocardiographie experte pour affiner le pronostic.
- Évaluation fonctionnelle globale : le score CVP
Pour évaluer le risque de défaillance cardiaque et de mort in utero, le Score de Profil Cardiovasculaire (CVP) est l’outil de référence. Ce score sur 10 points évalue cinq catégories :
- Hydrops : Absence (2 pts), épanchements localisés (1 pt) ou œdème cutané (0 pt).
- Taille du cœur : Rapport surface cardiaque/thoracique normal (2 pts) ou augmenté (0-1 pt).
- Fonction ventriculaire : Fraction de raccourcissement normale et absence de régurgitation valvulaire sévère.
- Doppler veineux : Flux normal dans la veine ombilicale et le canal d’Arantius.
- Doppler artériel : Présence d’un flux diastolique dans l’artère ombilicale.
Un score CVP $\le$ 7 est prédictif d’une augmentation significative de la mortalité périnatale et de la nécessité d’une intervention urgente. Une dégradation progressive de ce score lors d’un suivi sériel est un indicateur fort de compromis cardiovasculaire imminent.
III. Facteurs pronostiques des cardiopathies spécifiques
- Sténose aortique (SA) et évolution vers l’hypoplasie du cœur gauche
La SA critique présente un défi pronostique majeur : l’évolution in utero vers un syndrome d’hypoplasie du cœur gauche (SHCG). Les critères de sélection pour une valvuloplastie aortique fœtale (VAF) incluent une dysfonction ventriculaire gauche, un flux rétrograde dans l’arche aortique et un flux restreint à travers le foramen ovale.
Des modèles récents montrent que le rapport de longueur ventricule droit/ventricule gauche (RV/LV) associé à la vélocité de la régurgitation mitrale (MR-Vmax) prédit avec une haute sensibilité le succès d’une circulation biventriculaire (BV) après la naissance. Un rapport RV/LV < 1,094 est un excellent indicateur pour un devenir BV. Cependant, malgré le succès technique de la VAF (réussi dans plus de 80 % des cas), le taux global de circulation BV à un an reste d’environ 46 à 52 %, soulignant la sévérité de ces lésions.
- Cardiopathies univentriculaires et conotroncales
Dans les cœurs de type ventricule unique, la dominance du ventricule droit, la présence d’anomalies extracardiaques et un faible poids de naissance sont des facteurs de risque de mortalité précoce. Pour la Tétralogie de Fallot (TOF), un Z-score de la valve pulmonaire inférieur à -3 ou -5 et un flux rétrograde dans le canal artériel prédisent une dépendance ductale à la naissance, nécessitant des prostaglandines.
- Transposition des gros vaisseaux (TGA)
Le pronostic néonatal immédiat dépend de la perméabilité du foramen ovale. Des signes échographiques tels qu’un angle du septum primum < 30° ou un septum hypermobile suggèrent une restriction imminente, imposant une naissance dans un centre capable de réaliser une septostomie atriale (BAS) en urgence.
- L’axe cœur-cerveau : un nouveau paradigme pronostique
L’une des avancées les plus significatives concerne l’impact des anomalies cardiaques sur le développement cérébral in utero. Les CC complexes perturbent la perfusion et l’oxygénation cérébrale, entraînant des retards de maturation détectables par IRM fœtale.
- Hémodynamique cérébrale : Le flux dans la veine cave supérieure (SVC) est utilisé comme proxy du débit sanguin cérébral. Une réduction du flux SVC ou de la délivrance d’oxygène au cerveau (CDO2) est fortement corrélée à des scores cognitifs, linguistiques et moteurs plus faibles à 18 mois.
- Métabolisme cérébral : La spectroscopie par résonance magnétique (SRM) révèle que les fœtus porteurs de CC présentent des niveaux de lactate cérébral élevés (marqueur d’anaérobiose) et un rapport NAA/Choline abaissé (signe de retard de myelination) dès le troisième trimestre. Ces anomalies métaboliques sont associées à une augmentation du risque de décès avant la sortie de l’hôpital.
- Planification de la naissance et niveaux de soins
La synthèse de tous ces paramètres permet de stratifier les fœtus par Niveaux de Soins (LOC) :
- LOC 2 : Cardiopathies stables nécessitant une évaluation ou des prostaglandines à la naissance (ex: coarctation, SA sévère).
- LOC 4 : Cas critiques exigeant une équipe de réanimation cardiaque en salle de naissance et une intervention immédiate (ex: TGA avec septum intact, SHCG avec septum restrictif, arythmies incontrôlées avec hydrops).
Conclusion
L’évaluation pronostique moderne ne se limite plus à la survie, mais intègre la protection cérébrale et la planification multidisciplinaire (“Cardio-obstetrics team”). Si des outils comme le score CVP et les mesures de flux par IRM/CMR permettent aujourd’hui de mieux identifier les fœtus à haut risque, la gestion des cardiopathies les plus complexes comme la SA critique reste un défi où le conseil parental doit balancer les chances de succès d’une réparation biventriculaire face aux risques de morbidité neuro-développementale à long terme.
Conseil anténatal
Pr Yves Ville

L’objectif principal du conseil est de fournir aux parents une information précise, de définir le pronostic, de discuter des options de prise en charge et de faciliter une prise de décision partagée.
- L’Équipe Pluridisciplinaire : La « Cardio-Obstetrics Team »
La gestion des anomalies cardiaques fœtales repose sur une collaboration étroite entre divers spécialistes au sein d’une équipe de cardio-obstétrique. Cette équipe comprend idéalement des spécialistes en médecine fœtale (MFM), des cardiologues pédiatres ou fœtaux, des néonatologistes et, selon les cas, des chirurgiens cardiaques, des électrophysiologistes ou des conseillers en génétique. Une communication ouverte au sein de cette équipe est cruciale pour élaborer un plan de naissance optimal et cohérent. Le conseil doit être offert rapidement après l’échocardiographie diagnostique, idéalement le jour même, pour réduire l’incertitude parentale.
- Les Piliers de l’Information Médicale
Le conseil doit aborder trois aspects fondamentaux : le diagnostic anatomique, les implications génétiques et le pronostic à court et long terme.
- Précision Diagnostique et Évolution : Le praticien doit fournir un compte rendu honnête de la malformation, tout en expliquant les limites de l’imagerie (position fœtale, habitus maternel). Il est important d’informer les parents que certaines lésions peuvent progresser in utero, comme une sténose aortique évoluant vers un syndrome d’hypoplasie du cœur gauche.
- Évaluation Génétique : La découverte d’une CC doit systématiquement déclencher une discussion sur les tests génétiques, car 15 % à 30 % des fœtus avec une CC “isolée” présentent une anomalie chromosomique. Le risque varie selon la lésion : il est très élevé (46-73 %) pour un canal atrioventriculaire, mais faible pour une transposition des gros vaisseaux (TGV) isolée. La connaissance d’une cause génétique (ex: microdélétion 22q11.2) aide à prédire les résultats neurodéveloppementaux et à évaluer le risque de récurrence pour les grossesses futures.
- Pronostic et Devenir : Les parents doivent être informés de la survie post-opératoire et des morbidités potentielles. L’un des enjeux majeurs est le devenir neurodéveloppemental, souvent altéré dans les cardiopathies complexes en raison de perturbations de la perfusion cérébrale in utero.
III. Prise de Décision Partagée et Options de Prise en Charge
La prise de décision est un processus complexe où les valeurs des parents rencontrent l’expertise médicale.
- Interventions in utero : Dans des cas sélectionnés, des options comme la valvuloplastie aortique fœtale (VAF) peuvent être proposées pour tenter de préserver une circulation biventriculaire. Le conseil doit alors inclure les risques de mort fœtale liés à la procédure.
- Traitement des arythmies : Pour les tachycardies ou les blocs auriculoventriculaires (BAV) complets, le traitement transplacentaire maternel peut être discuté, en pesant les bénéfices fœtaux face aux risques de toxicité pour la mère.
- Interruption de Grossesse (IMG) et Soins Palliatifs : Pour les cardiopathies les plus sévères avec un pronostic sombre, l’IMG ou les soins palliatifs néonataux doivent être abordés comme des options valables. En France, l’IMG peut être acceptée jusqu’au terme pour des pathologies d’une particulière gravité.
- Planification de la Naissance et Stratification du Risque
Le conseil anténatal aboutit à la planification logistique de l’accouchement, basée sur la stratification de la sévérité de la lésion par niveaux de soins (LOC).
- LOC 1 et 2 : Pour les lésions simples ou ducto-dépendantes stables (ex: coarctation), une naissance dans un hôpital local avec accès à un néonatologiste et aux prostaglandines peut suffire.
- LOC 3 et 4 : Pour les cardiopathies critiques nécessitant une intervention immédiate en salle de naissance (ex: TGV avec septum intact, BAV avec hydrops), l’accouchement doit impérativement avoir lieu dans un centre de chirurgie cardiaque avec une équipe de réanimation spécialisée.
- Mode d’accouchement : Il est généralement dicté par des indications obstétricales plutôt que par la cardiopathie elle-même, bien qu’une césarienne programmée puisse faciliter la coordination des équipes dans les cas de niveau 4.
- Soutien Psychosocial et Techniques de Communication
Le diagnostic d’une anomalie cardiaque fœtale provoque un stress, une anxiété et un risque de dépression maternelle significatifs. L’évaluation de l’état émotionnel de la famille est une composante essentielle du conseil.
Les cliniciens doivent adapter leur langage au niveau de compréhension de la famille. L’utilisation de supports visuels, tels que des schémas du cœur normal comparé à la malformation, de vidéos, ou la visite de l’unité de soins intensifs cardiaques, peut grandement aider à la compréhension. Il est souvent nécessaire de répéter les informations sur plusieurs visites pour assurer une assimilation complète. Les parents apprécient également d’être orientés vers des sites web sécurisés ou des groupes de soutien.
Conclusion
Le conseil anténatal en cardiologie fœtale est bien plus qu’une simple transmission d’informations médicales. C’est une démarche d’accompagnement qui intègre la médecine de précision (génétique, imagerie avancée) à une approche centrée sur la famille. En identifiant précisément les fœtus à haut risque et en planifiant les interventions salvatrices dès la salle de naissance, cette approche systématique transforme radicalement le pronostic vital et fonctionnel des enfants nés avec une cardiopathie congénitale.
- Épidémiologie et Stratégie de Dépistage
- Prévalence : Les cardiopathies congénitales (CC) touchent environ 1 % des naissances vivantes (6 à 12 pour 1 000).
- Population concernée : Environ 90 % des CC surviennent dans la population à bas risque, sans aucun facteur prédisposant identifiable.
- Protocole standard : Le dépistage repose sur cinq coupes axiales transversales essentielles (situs abdominal, 4 cavités, LVOT, RVOT et 3VT).
- Taux de détection : L’approche par cinq coupes permet d’identifier jusqu’à 90 % des CC majeures contre seulement 50 % avec la seule coupe des 4 cavités.
- Signes Échographiques et Marqueurs Précoces
- Marqueurs du 1er trimestre : Une clarté nucale (CN) $\ge$ 3,5 mm, une régurgitation tricuspide ou une onde ‘a’ inversée dans le canal d’Arantius (DV) sont des signes d’alerte majeurs.
- Situs et Axe : L’apex cardiaque doit être orienté à gauche avec un axe de 45° ± 20°.
- Coupe des 3 vaisseaux et trachée (3VT) : Elle est cruciale pour détecter les anomalies de l’arc. Un “Signe du U” indique un arc aortique droit ou un anneau vasculaire, au lieu du “V” normal.
- Croisement des vaisseaux : L’absence de croisement orthogonal des gros vaisseaux (vaisseaux parallèles) est la signature de la transposition des gros vaisseaux (TGA).
III. Évaluation Fonctionnelle et Hémodynamique
- Score de Profil Cardiovasculaire (CVP) : C’est l’outil pronostique de référence sur 10 points. Un score $\le$ 7 est prédictif d’une morbidité et d’une mortalité périnatale élevées.
- Défaillance cardiaque : Elle se manifeste par une cardiomégalie, une dysfonction systolo-diastolique et, à un stade terminal, par des pulsations dans la veine ombilicale.
- Indice MPI (Tei) : Il permet d’évaluer la fonction cardiaque globale indépendamment de la géométrie ventriculaire.
- Génétique des Cardiopathies
- Rendement diagnostique : Le séquençage de l’exome (ES) apporte un diagnostic supplémentaire moyen de 31 % en cas d’anomalies structurelles multiples et de CMA normal.
- Microdélétion 22q11.2 : Elle est fortement associée aux anomalies conotroncales (Fallot, interruption de l’arche) et s’accompagne souvent d’une agénésie du thymus.
- Mutations de novo : Elles représentent environ 10 % des CC sévères et sont souvent liées à des gènes modificateurs de la chromatine.
- Axe Cœur-Cerveau et Neurodéveloppement
- Hémodynamique cérébrale : Le mécanisme de “brain sparing” (redistribution vers le cerveau) est visible par une baisse de l’indice de pulsatilité de l’artère cérébrale moyenne.
- Débit SVC : Une réduction du flux dans la veine cave supérieure (SVC) est directement corrélée à des scores cognitifs et moteurs plus faibles à 18 mois.
- Métabolisme cérébral : L’IRM montre que les CC complexes sont associées à une baisse du rapport NAA/Choline et à une élévation du lactate cérébral, marqueur de risque de décès néonatal.
- Troubles du Rythme et de la Conduction
- Tachycardies (SVT) : Elles nécessitent souvent un traitement transplacentaire par Digoxine, Flécaïnide ou Sotalol pour éviter l’hydrops.
- Blocs Auriculo-Ventriculaires (BAV) : Souvent liés aux anticorps maternels anti-Ro/SSA, ils présentent un risque de mortalité élevé si la fréquence ventriculaire est < 55 bpm.
- Magnétocardiographie (fMCG) : Technologie supérieure à l’échographie pour diagnostiquer le syndrome du QT long et les torsades de pointes.
VII. Conseil Anténatal et Prise en Charge
- Niveaux de soins (LOC) : La naissance doit être planifiée selon la sévérité. Le niveau 4 (LOC 4) exige une équipe de réanimation cardiaque immédiate en salle de naissance.
- Interventions in utero : La valvuloplastie (FAV) pour sténose aortique critique permet d’obtenir une circulation biventriculaire dans environ 52 % des cas réussis techniquement.
- Législation : En France, l’interruption médicale de grossesse (IMG) peut être acceptée jusqu’au terme pour des pathologies d’une particulière gravité.
- Approche multidisciplinaire : Le conseil doit impliquer une équipe de “cardio-obstétrique” pour assurer une cohérence entre le diagnostic, le pronostic neurodéveloppemental et le plan de naissance.
Les CC touchent environ 1 % des naissances vivantes. Leur prévalence est estimée entre 5 et 12 pour 1 000 naissances selon les études.
Car environ 90 % des fœtus porteurs d’une CC naissent de mères sans aucun facteur de risque identifiable.
Le protocole standard comprend : le situs abdominal, la coupe des quatre cavités (4C), la voie d’éjection gauche (LVOT), la voie d’éjection droite (RVOT) et la coupe des trois vaisseaux et de la trachée (3VT).
La CN est jugée augmentée lorsqu’elle est supérieure au 95e/99e percentile ou ≥ 3,5 mm.
L’ES offre un rendement diagnostique supplémentaire moyen de 31 % chez les fœtus présentant des anomalies structurelles multiples et un CMA normal.
La microdélétion 22q11.2 (syndrome de DiGeorge) est fortement associée à la tétralogie de Fallot et aux anomalies de l’arc aortique.
Le cœur est normalement orienté à gauche avec un axe de 45° ± 20°.
Elle indique souvent la présence d’un arc aortique droit ou d’un anneau vasculaire, où les vaisseaux entourent la trachée au lieu de converger en “V” à sa gauche.
La visualisation du croisement orthogonal des deux voies d’éjection (l’aorte et l’artère pulmonaire) à leur origine
Oui, un examen expert entre 11 et 14 semaines permet de détecter plus de 50 % des CC majeures.
La présence d’une onde ‘a’ inversée ou absente (pendant la contraction auriculaire) est un marqueur de risque accru de CC.
Il faut respecter le principe ALARA et maintenir les indices thermique (TI) et mécanique (MI) ≤ 1,0
C’est un outil évaluant la fonction globale (systolo-diastolique) en mesurant les temps de contraction/relaxation isovolumique et le temps d’éjection.
Un flux anormal dans le DV ou des pulsations dans la veine ombilicale indiquent une augmentation de la pression veineuse centrale et une défaillance cardiaque droite.
C’est une redistribution du débit cardiaque vers le cerveau, visible par une baisse de l’indice de pulsatilité (PI) dans l’artère cérébrale moyenne.
La présence d’un flux rétrograde (inversé) dans l’isthme aortique ou le canal artériel.
Une large communication interventriculaire (CIV) avec une aorte “à cheval” sur le septum.
Par l’absence de la croix du cœur normale et la présence d’une valve unique commune remplissant les deux ventricules (image en “Y” au Doppler).
Un ventricule gauche minuscule et, en coupe 3VT, un flux rétrograde (bleu) dans un arc aortique très étroit.
Une disproportion ventriculaire avec un ventricule droit significativement plus large que le gauche.
Une vélocité élevée (MR-Vmax) est un prédicteur de la capacité du ventricule gauche à maintenir une circulation biventriculaire après traitement.
L’hydrops est un signe de décompensation terminale associé à une mortalité périnatale très élevée.
Elle permet un enregistrement continu type Holter et analyse précisément la repolarisation (intervalle QT), souvent invisible à l’échographie.
Devant une bradycardie sinusale inexpliquée ou des épisodes de torsades de pointes.
Le Flécaïnide est de plus en plus utilisé, souvent en association avec la Digoxine en cas d’hydrops.
La présence d’anticorps maternels anti-Ro/SSA et anti-La/SSB.
Une fréquence inférieure à 55 bpm est associée à un risque élevé de défaillance cardiaque.
C’est un score sur 10 points évaluant l’hydrops, la taille du cœur, la fonction ventriculaire et les flux Doppler artériels/veineux.
Un score ≤ 7 est prédictif d’une morbidité et d’une mortalité accrues.
La détection de lactate cérébral élevé au troisième trimestre par spectroscopie.
En mesurant le débit dans la veine cave supérieure (SVC), qui sert de proxy pour le flux cérébral.
Un groupe multidisciplinaire incluant des experts en médecine fœtale, des cardiologues pédiatres, des néonatologistes et des conseillers en génétique.
Il désigne les cardiopathies critiques nécessitant une naissance dans un centre spécialisé avec une intervention immédiate en salle de naissance.
L’IMG peut être acceptée jusqu’au terme si la pathologie est d’une particulière gravité, indépendamment du succès possible d’interventions fœtales.
Généralement non ; la voie basse est privilégiée sauf indications obstétricales ou cas de LOC 4 nécessitant une coordination extrême.
Le terme complet est défini entre 39 0/7 et 40 6/7 semaines de gestation.
C’est une variante vasculaire qui passe derrière la trachée. Isolée, elle est souvent bénigne mais peut être un marqueur mineur de trisomie 21.
Tenter de prévenir l’évolution d’une sténose aortique vers un ventricule gauche non fonctionnel (SHCG) et préserver une circulation biventriculaire.
Il s’évalue dans la coupe 3VT. Son absence est un signe fort de microdélétion 22q11.2.
Améliorer la survie et le devenir neuro-développemental en planifiant une prise en charge adaptée dès la naissance.