Demo

Demo
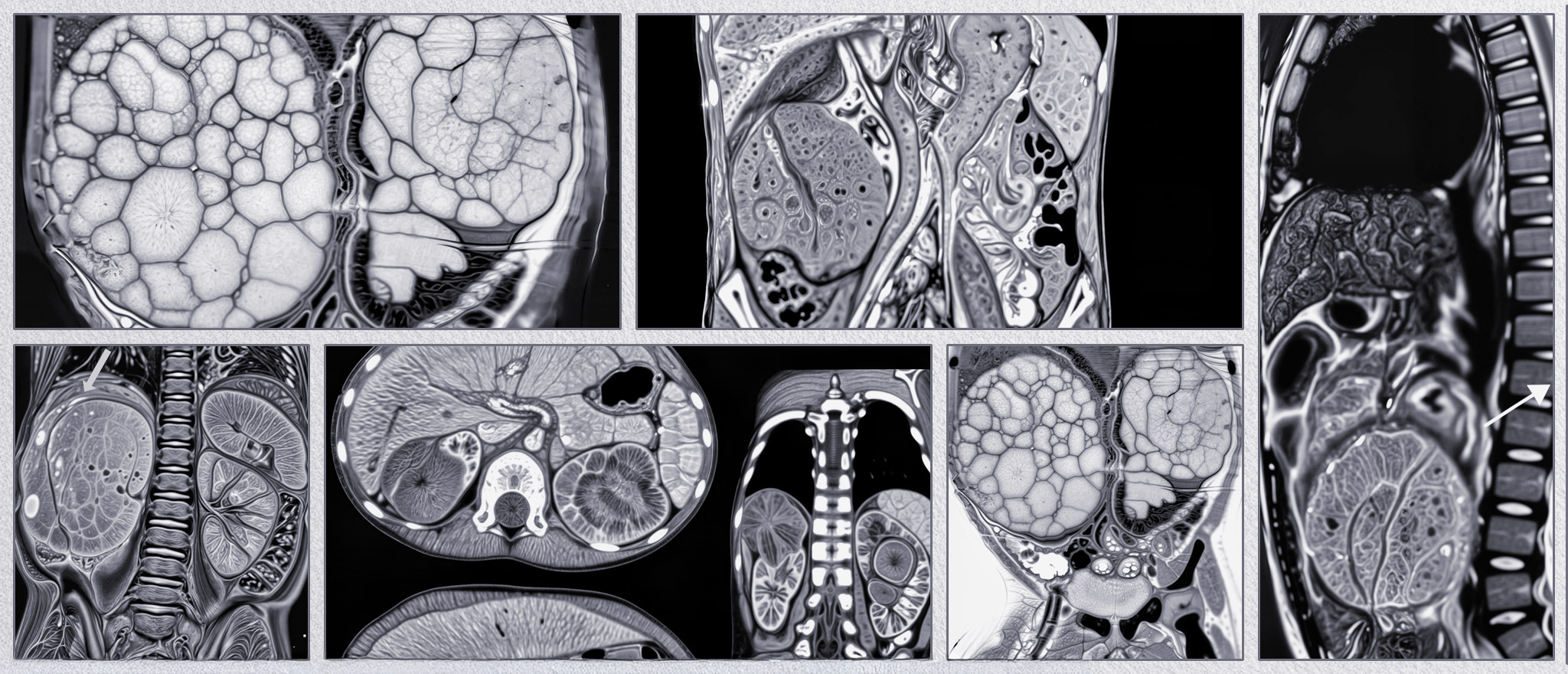
BEFORE
AFTER
Faire glisser le slider
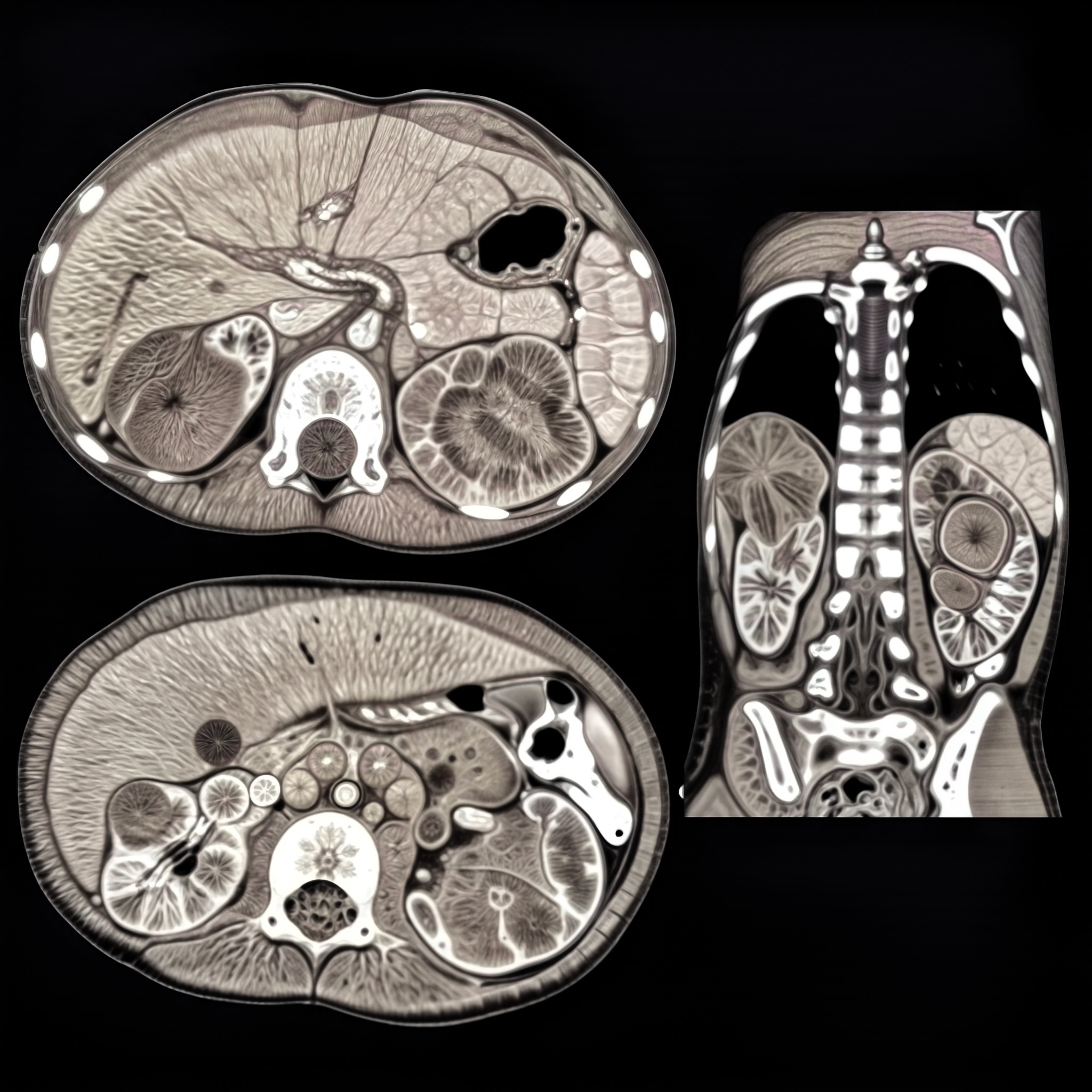
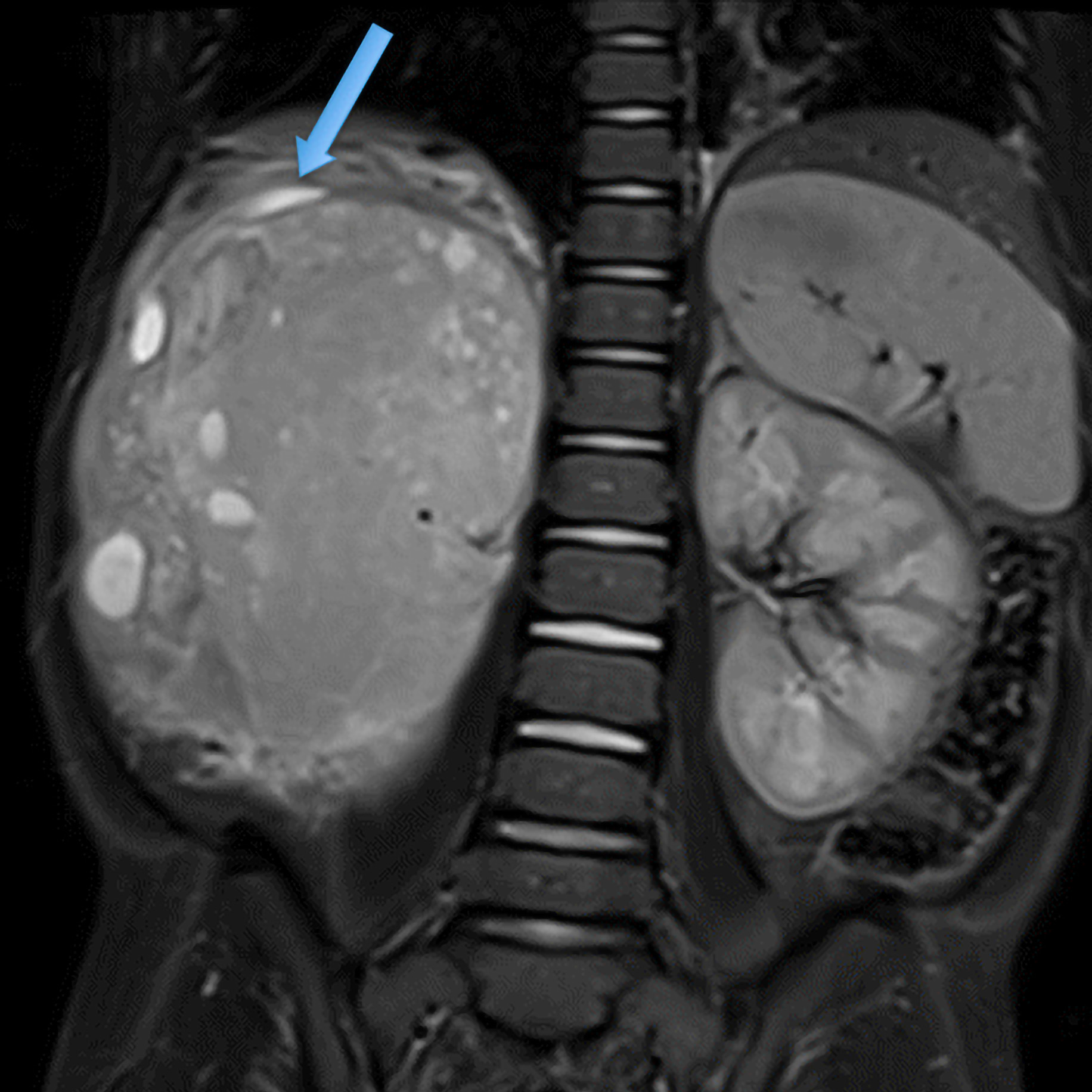
BEFORE
AFTER
Faire glisser le slider
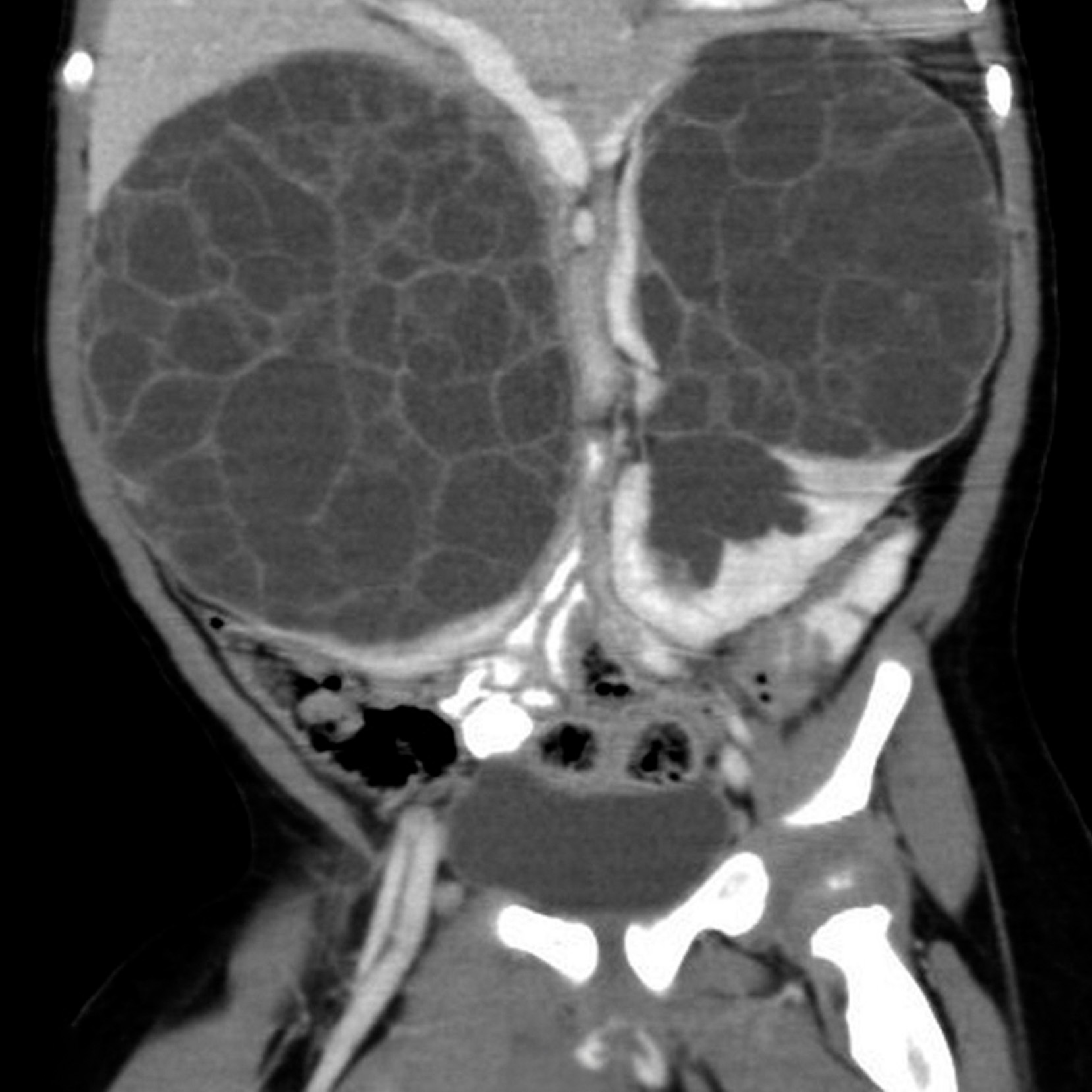
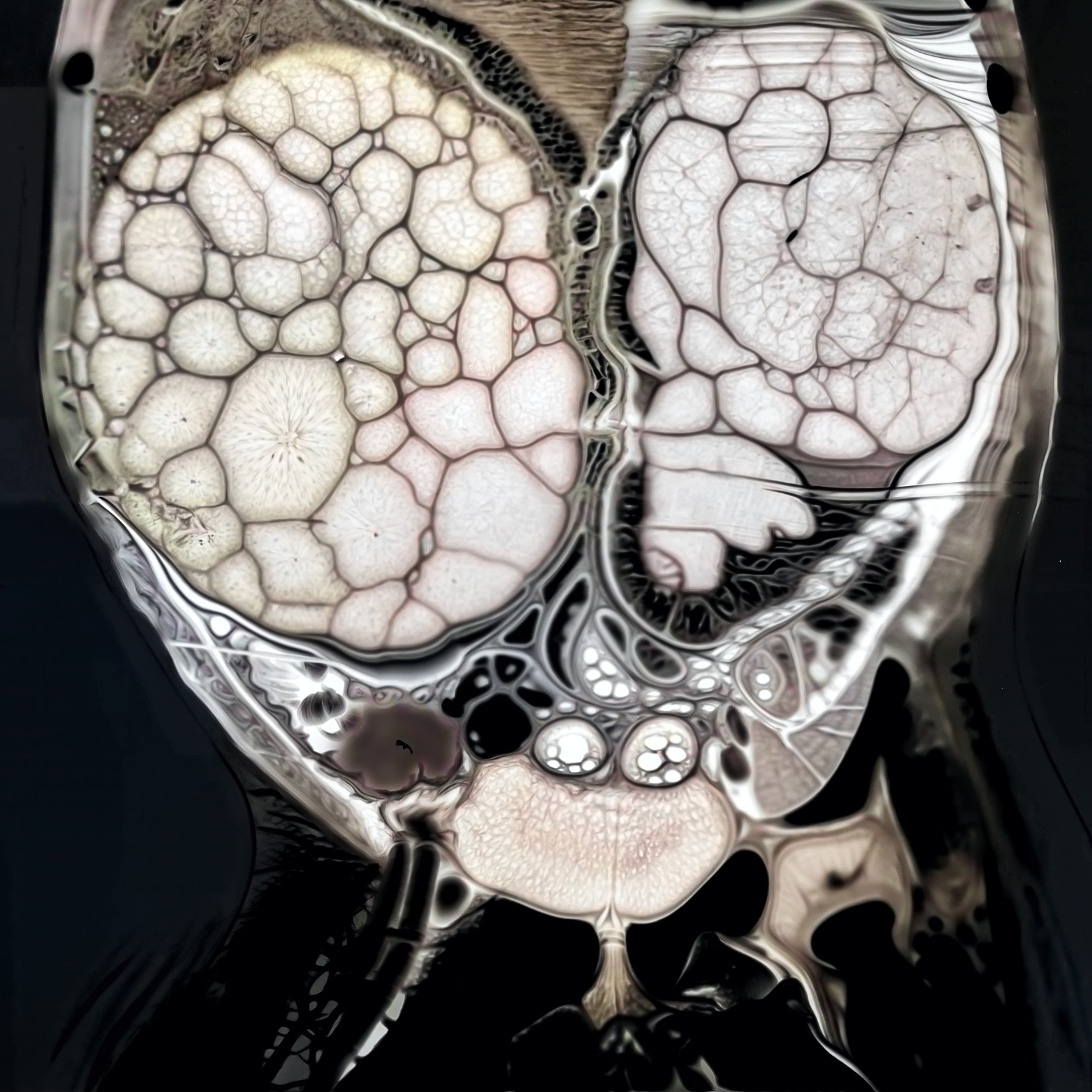
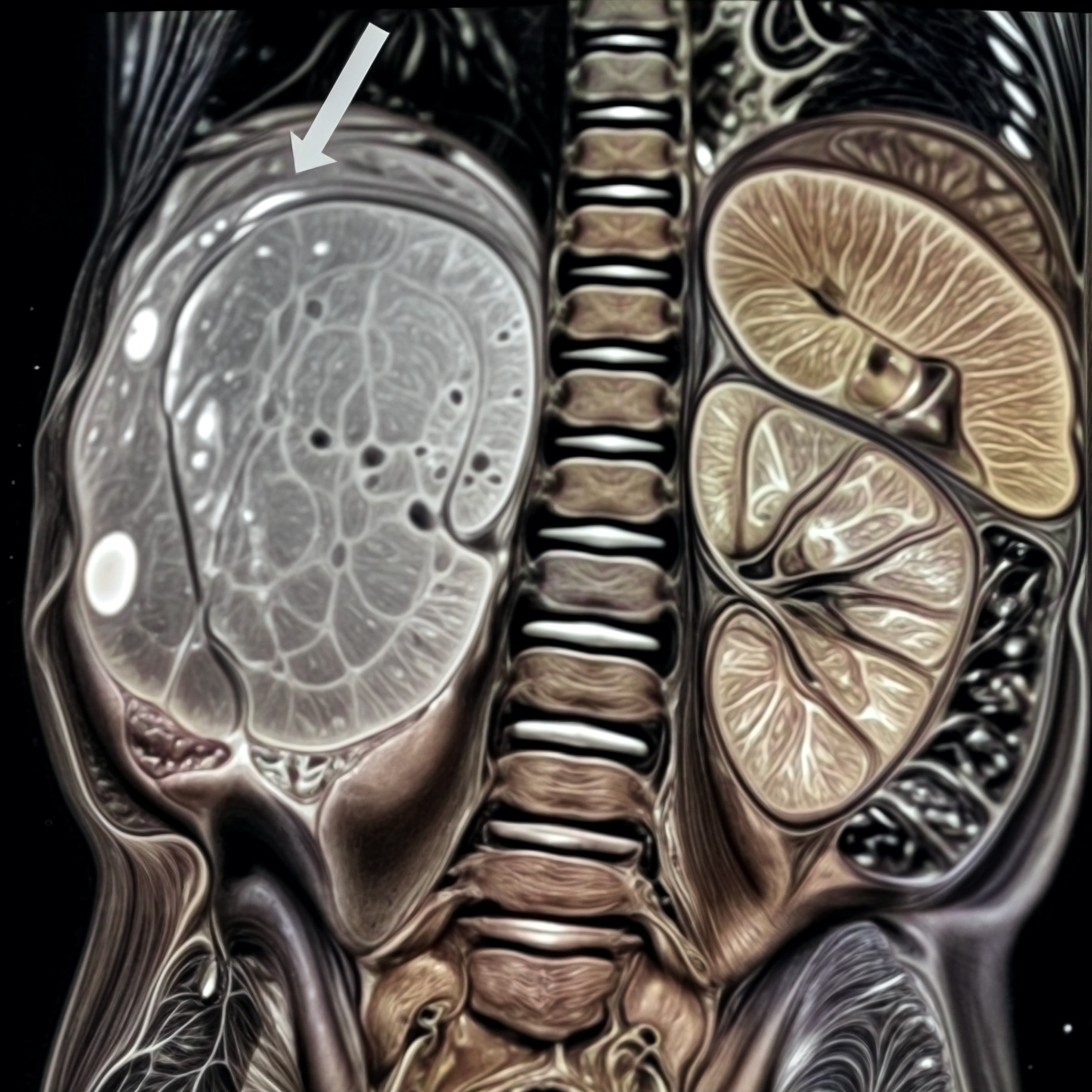
BEFORE
AFTER
Faire glisser le slider
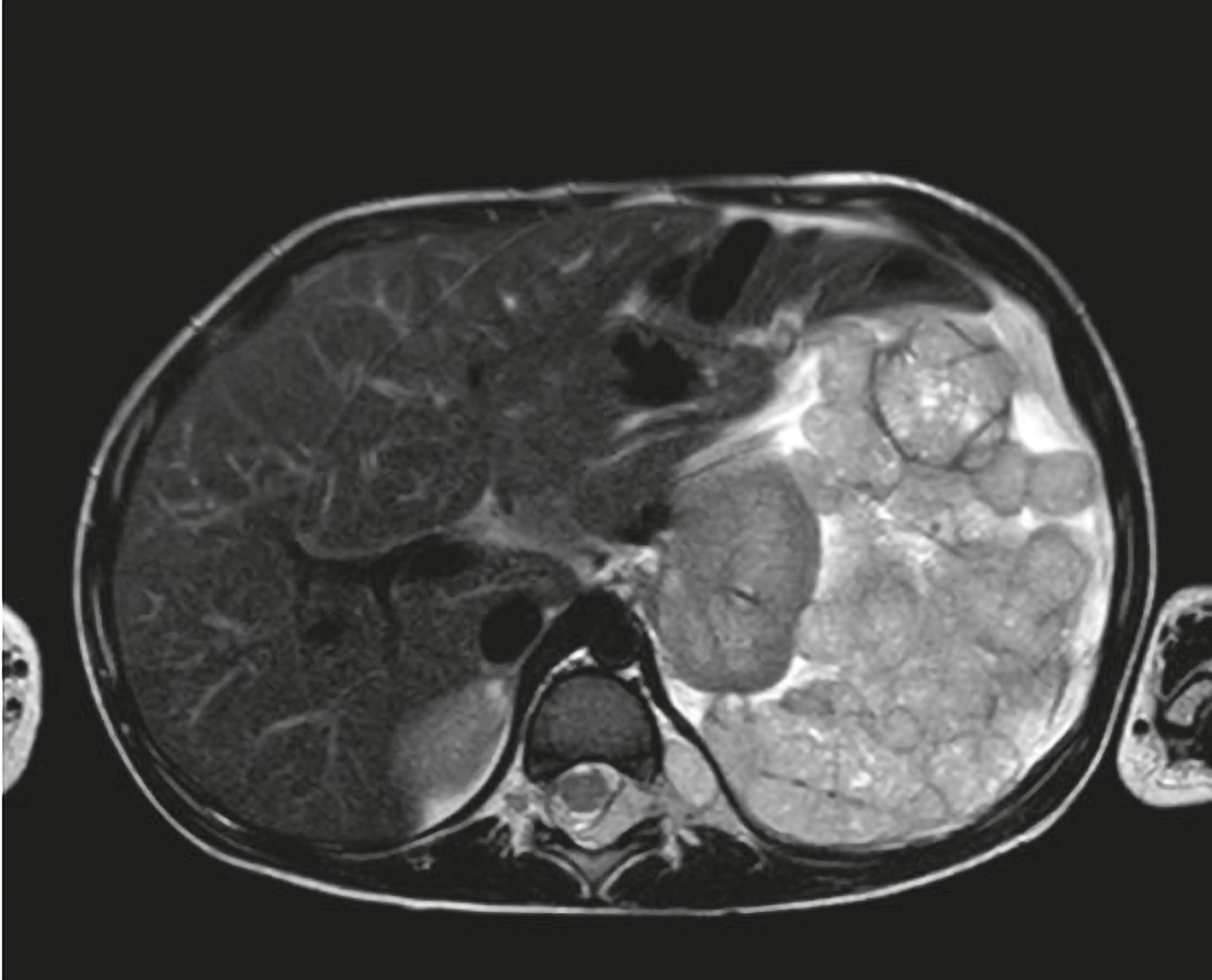
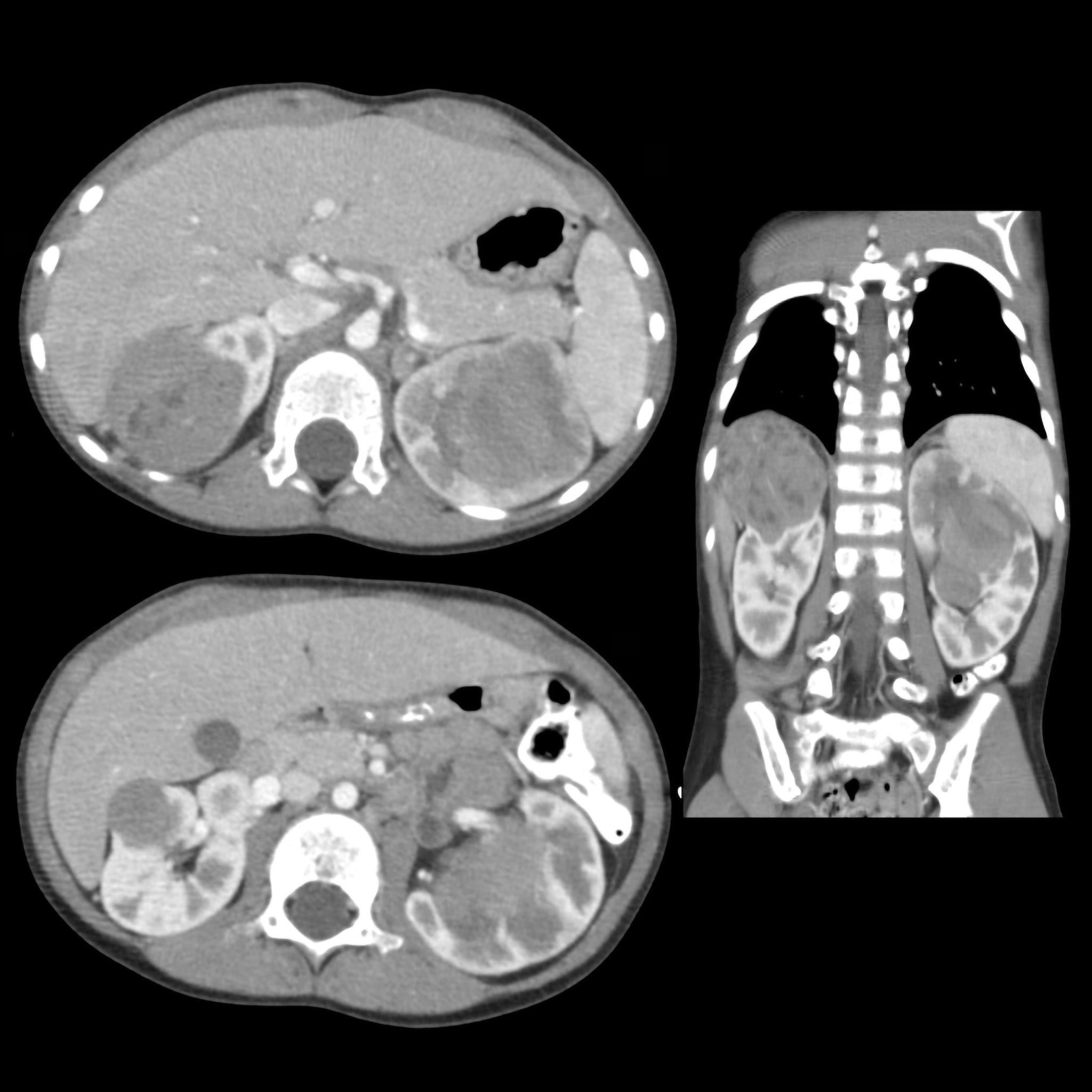
BEFORE
AFTER
Faire glisser le slider
Bibliographie



- Overview of WT1: Biology and Structure
WT1 (Wilms Tumor 1) is a zinc finger transcription factor encoded on chromosome 11p13, originally described as a tumor suppressor in pediatric nephroblastoma. However, its function is far more complex: depending on the cellular context, WT1 can act as an oncogene or a tumor suppressor. It regulates gene transcription, cellular differentiation, and organogenesis during development, and maintains homeostasis in adult tissues.
WT1 produces multiple isoforms (+KTS, –KTS) through alternative splicing, each playing distinct roles in gene expression and tumor biology. Its expression is typically restricted to embryonic and select adult tissues like kidney podocytes, but is aberrantly upregulated in a wide array of cancers.
- Hematologic Malignancies and WT1
a) Acute Myeloid Leukemia (AML)
WT1 is mutated in ~15% of AML patients and often co-mutated with FLT3-ITD or NPM1. These mutations influence the Wnt/β-catenin, TET2, and MEG3 pathways, promote leukemogenesis, and impact response to chemotherapy. WT1 overexpression enhances stemness, self-renewal, and resistance to apoptosis.
b) Myelodysplastic Syndromes (MDS)
WT1 is overexpressed in ~50% of MDS patients. It is associated with poor prognosis and disease progression. WT1 interacts synergistically with FLT3 mutations, contributing to transformation to AML.
c) T-cell Acute Lymphoblastic Leukemia (T-ALL)
WT1 mutations (~10% of T-ALL cases) activate the IL7R-JAK-STAT pathway and impair TP53-mediated DNA damage response. Co-occurrence with NOTCH1/FBXW7/FLT3 mutations suggests an oncogenic synergy.
d) Chronic Myeloid Leukemia (CML)
WT1 expression is increased in advanced CML via BCR-ABL1 activation. It contributes to resistance against tyrosine kinase inhibitors like imatinib by interfering with apoptosis and gene regulation through the WT1/ZNF224/c-Myc axis.
e) Acute Promyelocytic Leukemia (APL)
WT1 interacts with the PML-RARA fusion protein, altering differentiation. Noncoding mutations in WT1 may also contribute to leukemogenesis.
- WT1 in Solid Tumors
a) Lung Cancer
WT1 promotes VEGF isoform expression and angiogenesis. It may serve as a target to reverse resistance to EGFR inhibitors in non-small-cell lung cancer (NSCLC).
b) Breast Cancer
WT1 isoforms have distinct roles in ER-positive and triple-negative breast cancer. They affect ERα signaling, promote EMT and metastasis, and may predict resistance to hormone therapy. WT1-targeted vaccines in combination with endocrine therapy show promising early results.
c) Neuroblastoma
In neuroblastoma, WT1 often functions as a tumor suppressor. It promotes differentiation through inhibition of PI3K/Akt and MAPK/ERK pathways. However, high WT1 is paradoxically associated with poor prognosis in non-MYCN-amplified tumors.
d) Prostate Cancer
WT1 downregulates E-cadherin, facilitating migration and metastasis. It also modulates VEGF transcription and androgen receptor activity, influencing hormone resistance.
e) Hepatocellular and Pancreatic Cancer
WT1 influences Wnt/β-catenin, JAK2/STAT3, and NF-κB pathways. It promotes tumorigenesis, invasion, and modulates immune escape mechanisms. In pancreatic ductal adenocarcinoma, it interacts with STAT3 and miR-216a/KRT7, facilitating metastasis.
f) Ovarian Cancer
WT1 is highly expressed in serous subtypes and promotes EMT via ERK1/2 and E-calmodulin pathways. It is involved in mesenchymal–epithelial transitions (MET) and cancer stem cell formation through interaction with EZH2.
g) Astrocytomas/Glioblastoma
WT1 contributes to glioblastoma aggressiveness and poor prognosis. Its inhibition enhances radiosensitivity, and WT1 vaccines elicit cytotoxic immune responses in glioma patients.
- WT1 as a Therapeutic Target
a) Chemical Inhibitors
Agents like 17-AAG (Hsp90 inhibitor), TMPyP4 (G-quadruplex stabilizer), and daunorubicin reduce WT1 expression. These agents inhibit tumor growth and increase chemosensitivity in WT1-overexpressing malignancies.
b) Natural Products
Compounds such as bufalin, shikonin, aloin, curcumin, and resveratrol suppress WT1 expression and modulate apoptosis pathways. Marine-derived molecules also show potential in WT1 inhibition.
c) Kinase Modulators
GSK3β affects WT1 stability and degradation. Dasatinib is proposed as a modulator to reduce cytokine release syndrome in combination therapies.
- Immunotherapy Strategies
- Peptide Vaccines: WT1-derived peptides induce T-cell and humoral responses; trials show tolerability and clinical activity in AML and glioblastoma.
- Dendritic Cell Vaccines: Boost CD8+ T-cell responses; combination with chemotherapy improves remission rates.
- Bispecific Antibodies: T-cell engaging antibodies (e.g., ESK1-BiTE) target WT1 in HLA-A*02:01 context.
- TCR Gene Therapy: Autologous T cells engineered to express WT1-specific TCRs show persistence and safety in early-phase trials.
Conclusion and Future Prospects
WT1 is a compelling, dual-role protein in cancer biology. It functions as a context-dependent oncogene or tumor suppressor, modulating multiple pathways in both hematologic and solid tumors. Its overexpression correlates with treatment resistance and poor prognosis.
Targeting WT1 is now a key area of translational research, with therapeutic strategies spanning small molecules, natural products, and immunotherapies. Though many approaches remain in preclinical or early clinical stages, the diversity of mechanisms through which WT1 drives cancer supports its status as a pan-cancer therapeutic target.
Comprehensive Summary
This narrative review explores the evolution of personalized medicine in the context of Wilms tumor (WT), the most common renal malignancy in children. It synthesizes recent advancements in molecular diagnostics, risk stratification, and therapeutic innovations, offering a roadmap for the development of individualized care pathways in pediatric oncology.
- Background and Rationale
Wilms tumor accounts for roughly 7% of all childhood cancers, with a high survival rate (>90%) for localized disease in developed countries. However, variability in outcomes persists due to biological heterogeneity, socioeconomic disparities, and treatment-related toxicity. The shift toward risk-adapted and genetically-informed strategies is positioned as key to improving outcomes while minimizing late effects.
- Genetic and Molecular Foundations
Key genetic players such as WT1, CTNNB1, SIX1, MYCN, and WTX are central to WT pathogenesis. The review emphasizes that:
- WT1 mutations are highly predictive of prognosis, especially in bilateral disease.
- SIX2 expression, DNA methylation profiles, and circulating miRNAs (e.g., miR-144-3p) provide non-invasive biomarkers.
- Mutations in DIS3 and TERT are noted in relapsed WT, underlining the evolution of tumor biology over time.
- Diagnostics: Beyond Morphology
The authors advocate for integrating imaging, molecular, and epigenetic data to enable early detection and treatment personalization:
- Ultrasound, MRI, and CT remain standard imaging tools, but now enhanced with radiomics and 3D segmentation.
- Liquid biopsies, including ddPCR for PIK3CA mutations in urine/plasma, offer a minimally invasive route to real-time monitoring.
- Genomic and transcriptomic tools provide detailed tumor profiling, now entering routine clinical pipelines.
- Risk Stratification and Prognostic Modeling
The review underscores a paradigm shift from stage-based to biomarker-based stratification:
- Tumor volume, while still a classical metric, gains predictive power when integrated with molecular profiles.
- Nomograms using multi-omic and clinical data help forecast recurrence, survival, and therapy response.
- Machine learning models incorporating radiomics, genomics, and clinical data are under development to support decision-making.
- Therapeutic Innovations and Personalization
- a) Standard Therapy with Personalization
- Traditional agents (vincristine, actinomycin D, doxorubicin) remain effective, but dose and duration are increasingly adjusted based on molecular risk.
- Nephron-sparing surgery and preoperative chemotherapy are optimized using volumetric and genetic data.
- b) Targeted Therapies
- HIF-2α inhibitors and IGF-1R pathway blockers are in trials for treatment-resistant cases.
- Targeting the Wnt/β-catenin and AKT pathways is a key focus.
- c) Immunotherapy
- WT1 peptide vaccines, immune checkpoint inhibitors (PD-1/PD-L1), and CAR-T cells are emerging options, especially for relapsed/refractory WT.
- Combining WT1 vaccines with HLA-matched peptides enhances immune targeting with early evidence of improved outcomes.
- d) Genetic and Epigenetic Interventions
- Techniques such as CRISPR-Cas9, RNA interference, and methylation modulators are in early-phase development.
- These strategies aim to reverse tumor-promoting gene expression and reduce resistance mechanisms.
- Radiomics and Advanced Imaging
The review details how radiomic analysis and AI-assisted imaging can predict histological subtypes, therapeutic response, and relapse risk. Coupled with genomic data, they provide the basis for integrative predictive models that will soon guide both surgical planning and post-treatment surveillance.
- Global Collaboration and Equity in Care
- The authors call for international cooperation to validate predictive models and ensure equitable access to genomic testing and targeted therapies.
- Socioeconomic disparities must be addressed to democratize personalized medicine, particularly in resource-limited settings.
- Future Directions and Framework Proposal
The authors propose a multi-dimensional predictive framework based on:
- Next-generation sequencing (NGS) for WT1/CTNNB1/WTX
- Epigenomic profiling for methylation and histone changes
- Transcriptomic analysis (e.g., RNA-seq)
- Radiomic features from diffusion-weighted MRI
- Integrated AI models for real-time decision support
- Clinical teams trained in data interpretation and continuous feedback updating from registries
Conclusion
This review establishes that the future of WT therapy lies in personalization. By integrating multi-omic diagnostics, predictive modeling, and innovative therapies, clinicians can move toward tailoring care to the individual child. These efforts promise not only higher survival, but also reduced toxicity, better quality of life, and long-term survivorship.
Comprehensive Summary
This retrospective audit analyzes 23 pediatric Wilms’ tumor (WT) cases managed at a single Indian tertiary-care center over a 3-year period (2021–2024). The study evaluates how SIOP Umbrella protocols were applied and adapted in a resource-limited setting, offering insights into protocol adherence, surgical approaches, tumor response, immunohistochemistry, and outcomes in unilateral and bilateral WT.
- Patient Demographics and Study Design
- Total patients: 23 (19 unilateral WT [uWT], 4 bilateral WT [bWT])
- Median age: 36 months
- Male:Female ratio: 2.3:1
- Data sources: Medical records, imaging, core-needle biopsy (CNB), histopathology, immunohistochemistry (IHC)
- Ethical approval obtained; study focused only on de novo cases treated in-house
- Diagnostics and Protocol Deviations
- CNB performed in 22/23 patients pre-therapy, including IHC for WT1, p53, and Ki-67
- Despite SIOP-RTSG discouraging routine CNB, it was performed routinely here due to the higher incidence of non-Wilms renal tumors (15–20%) in South Asia
- All patients, except one neonate, received neoadjuvant chemotherapy (NACT)
- Tumor volumetry (via CECT): median pre-NACT tumor volume = 1023 mL, post-NACT = 612 mL
- Tumor shrinkage seen in 61%, stable/increased in 39% – volume reduction was not predictive of outcome
- Surgical Management
- Nephroureterectomy in 17 renal units
- Nephron-sparing surgery (NSS) in 10 renal units (including in non-syndromic uWT and multifocal disease)
- NSS sometimes performed against Umbrella guidelines, particularly in tumors >300 mL
- NSS associated with better postoperative renal function, lower hypertension, and preserved renal mass
- No intraoperative tumor rupture (IOTR) occurred, despite several large tumors
- Histopathology and Risk Stratification
- Intermediate risk histology: 21 patients (mixed, stromal, regressive, focal anaplasia)
- High risk: 2 patients (diffuse anaplasia, blastemal type)
- IHC findings:
- WT1 positivity in 21/23
- p53 mutations in 7/23
- High Ki-67 (>70%) correlated with chemoresistance and poor prognosis
- Low Ki-67 + p53 mutation often predicted poor NACT response
- Chemotherapy, Radiation, and Supportive Care
- Adjuvant chemotherapy completed in 18 patients, 2 on treatment
- Flank radiation used in 6 patients
- Multidisciplinary care included free lodging, nutrition, and counseling (“Home Away from Home” model)
- In-patient chemotherapy reduced abandonment risk
- Despite lacking in-house radiotherapy, radiation was started within 10 days post-op via national network
- Recurrences and Metastases
- Four patients had metastatic WT at diagnosis: 2 with pulmonary, 2 with liver metastasis
- Pulmonary metastases were managed with chemotherapy ± bilateral metastatectomy
- Liver metastasis had worse outcomes, including one metachronous recurrence post-treatment
- Two recurrences occurred locally (renal bed); both patients survived after multimodal therapy
- Outcomes and Survival Analysis
- 1-year overall survival (OS): 84%
- 1-year event-free survival (EFS): 76%
- 2-year OS: 77%
- 2-year EFS: 76%
- For localized uWT (Stages I–III), 2-year OS and EFS reached 100% and 93%, respectively
- Outcomes for Stage IV and bWT remain poor, mirroring national data
- Discussion and Strategic Insights
- Switching from upfront surgery to SIOP-based neoadjuvant chemotherapy dramatically improved outcomes, particularly in reducing IOTR and relapses
- Challenges remain in identifying non-responders to NACT, particularly those with stromal or blastemal histology
- Systematic use of CNB and IHC helps in pre-treatment risk identification, albeit controversial
- The use of centralized support services, early multidisciplinary planning, and enhanced follow-up are crucial for optimal outcomes in LMICs
- Despite limitations, the center matched short-term outcomes from high-income countries for localized WT
Summary:
This comprehensive review synthesizes current knowledge on Wilms tumor (WT)—also called nephroblastoma—with an emphasis on its clinical profile, treatment paradigms, molecular markers, and research gaps. It provides a global perspective and addresses both standardized treatment advances and novel, personalized approaches, while also recognizing challenges faced in resource-limited settings.
- Clinical Features and Classification
Wilms tumor is the most common pediatric renal malignancy, accounting for over 90% of childhood kidney tumors. It typically occurs in children under 5 years old, and more than 90% of diagnoses are made before age 3.
- Histologic classification includes:
- Triphasic: blastemal, epithelial, and stromal elements (most common)
- Biphasic/Monophasic: less frequent, variable prognosis
- Bilateral tumors occur in 4–8% of cases and are often syndromic or associated with nephroblastomatosis.
- Syndromic associations: WAGR, Denys-Drash, Beckwith-Wiedemann
- Clinical presentation: painless abdominal mass, hematuria, hypertension, and occasionally congestive heart failure
- Molecular and Genetic Underpinnings
- Key gene mutations: WT1, WT2 (11p15), CTNNB1, TP53, WTX
- Loss of heterozygosity (LOH) at 1p, 16q, and gain of 1q are critical prognostic biomarkers
- Mutations in TP53 are linked to anaplastic histology and poor outcomes
- Epigenetic alterations: promoter methylation of genes like HIC1, RASSF1, and miRNA profiles (miR-21, miR-130b) serve as prognostic and predictive markers
- Diagnosis and Imaging
- First-line imaging: ultrasound, followed by CT or MRI
- Apparent diffusion coefficient (ADC) values on MRI correlate with histopathology post-chemotherapy
- Imaging is essential for:
- Detecting venous thrombus
- Determining tumor volume
- Assessing treatment response
- Current and Emerging Treatments
- a) Standard Protocols
- Based on COG and SIOP frameworks
- Treatment includes nephrectomy, chemotherapy, and sometimes radiotherapy
- Flank radiation is associated with a significantly higher rate of late secondary malignancies (up to 61%)
- b) Nephron-Sparing Surgery (NSS)
- NSS is increasingly used in selected cases (especially bilateral WT) and shows comparable outcomes with total nephrectomy for localized tumors
- c) Novel Therapeutics
- Targeted agents: PIK3R3 inhibitors (ZSTK474), HIF-1α inhibitors, BET inhibitors (e.g., BRD4)
- Immunotherapy: adoptive T-cell therapy, vaccines targeting WT1 antigens
- Multidrug combinations (e.g., vincristine + irinotecan + temozolomide + bevacizumab) are being trialed for relapsed disease
- Disparities in Global Outcomes
- High-income countries report >90% survival for localized WT
- Low- and middle-income countries (LMICs) face <50% survival due to:
- Late presentation
- Treatment abandonment
- Malnutrition
- International collaborations like the Wilms Tumor Africa Project are addressing these disparities through protocol harmonization
- Future Research Directions
- Large-scale genomic and epigenomic studies are required to better define:
- High-risk tumor biology
- Chemoresistance mechanisms
- Prognostic biomarkers (e.g., lncRNAs like SOX21-AS1)
- Tumor microenvironment and miRNAs are emerging as targets for therapy
- Rare variants like teratoid Wilms tumor warrant dedicated studies due to atypical biology
- Long-Term Outcomes and Surveillance
- 15% of patients experience recurrence, often with high mortality
- Late effects include:
- Second malignancies (especially after radiotherapy)
- Renal dysfunction
- Psychosocial complications
- Surveillance strategies include regular imaging and screening for complications
Conclusion
The review emphasizes the multifactorial nature of Wilms tumor involving histological, genetic, and environmental components. As survival improves, efforts must focus on risk stratification, reduction of treatment burden, and integration of precision medicine tools. Future success in WT care hinges on international cooperation, advanced molecular diagnostics, and the development of integrative predictive models.
Comprehensive Summary:
This review explores the evolution of prognostic factors in Wilms tumor (WT) management, emphasizing the integration of clinical, histological, and molecular markers to guide risk-adapted therapy. Through comparative analysis of the Children’s Oncology Group (COG) and International Society of Pediatric Oncology (SIOP) protocols, the article outlines how risk stratification has shaped personalized therapy, enhanced outcomes, and reduced treatment toxicity.
- Epidemiology and Background
Wilms tumor is the most common malignant renal tumor in children and the second most frequent extracranial solid tumor. It occurs in ~1 in 10,000 children and is diagnosed before the age of 5 in the majority of cases. Most tumors are unilateral, but 5–10% are bilateral, often associated with genetic syndromes like WAGR, Denys-Drash, and Beckwith-Wiedemann.
- Historical Risk Stratification
Originally, only tumor stage and histologic subtype were used to define risk and guide treatment. COG and SIOP have progressively expanded these criteria to incorporate tumor volume, genetic markers, response to therapy, and age at diagnosis.
- Key Prognostic Markers
a) Tumor Stage and Histology
- Still foundational, but now refined with additional criteria.
- Anaplastic histology remains a critical negative prognostic factor, requiring intensified chemotherapy and radiotherapy.
b) Loss of Heterozygosity (LOH) at 1p and 16q
- Associated with poorer prognosis and higher recurrence.
- Patients with LOH at either locus receive more aggressive chemotherapy.
c) Chromosome 1q Gain
- An emerging biomarker linked to adverse outcomes.
- Found in ~30% of patients; its integration in future trials is planned.
d) Lung Nodule Response
- In stage IV patients with pulmonary metastases, response to chemotherapy dictates whether whole-lung irradiation (WLI) can be safely omitted.
- 1q gain predicts poor EFS if WLI is omitted.
- COG vs. SIOP Strategies
COG Approach:
- Upfront nephrectomy enables early histological and molecular characterization.
- Intensifies treatment in patients with diffuse anaplasia, incomplete lung response, or LOH at 1p/16q.
- Recent trials: AREN0532, AREN0533, AREN0321.
SIOP Approach:
- Emphasizes preoperative chemotherapy to reduce intraoperative tumor rupture.
- Risk stratification includes volumetric and histologic response to therapy.
- UMBRELLA protocol: prospective global study aiming to integrate new molecular markers, standardize radiologic assessment, and personalize therapy further.
- Molecular Advances and Future Directions
- Circulating tumor DNA (ctDNA), microRNAs (miRNAs), and methylation profiling show promise as minimally invasive tools for diagnosis and monitoring.
- Machine learning models integrating genomic, radiologic, and clinical data are under development.
- Future trials (COG and SIOP) will use 1q gain status to tailor therapy: omitting or augmenting based on molecular risk.
Conclusion
The refinement of risk stratification in Wilms tumor has led to safer and more effective therapies, improving survival while reducing long-term toxicity. By incorporating clinical, histologic, and molecular markers, and leveraging treatment response data, clinicians can now offer truly personalized care. Ongoing and future research will focus on refining predictive models and incorporating liquid biopsy, genomic profiling, and AI-based stratification into routine clinical practice.
Extended Summary
This article presents a retrospective analysis of late adverse effects in survivors of Wilms’ tumor (WTS) treated between 1977 and 2023 at a single pediatric oncology center in Spain. With advances in treatment increasing survival rates above 90%, this study focuses on understanding the chronic health conditions that may arise long after therapy completion.
- Study Cohort and Methodology
- Sample size: 50 patients (25 males, 25 females)
- Age at diagnosis: Mean = 3.6 years; 86% diagnosed before age 5
- Tumor laterality: 85% unilateral, 12% bilateral, 1 extrarenal
- Treatment strategy: 94% followed ISPO (European) protocol; 1 patient treated per NWTSG (American) protocol
- Neoadjuvant chemotherapy: Administered to 90%
- Most common drugs: Actinomycin D and vincristine (78%), anthracyclines (28%)
- Surgery: 84% underwent total nephrectomy
- Radiotherapy: 34% received it, mostly postoperatively
- Adverse Effects – Overview by Organ System
a) Renal Disorders (46%)
- Most common sequela
- 11% developed chronic renal disease (CRD), GFR < 90 mL/min
- 25% experienced recurrent urinary infections, inflammation, or renal lithiasis
b) Cardiac Disorders (23%)
- Linked mostly to anthracyclines (e.g., doxorubicin)
- Manifestations: hypertension (13%), ventricular dysfunction (4%), septal hypertrophy, bicuspid aortic valve
c) Endocrine Disorders (26%)
- Predominantly subclinical hypothyroidism (13%) and obesity
d) Reproductive Disorders (13%)
- 4% of girls had diminished ovarian reserve after radiotherapy
- 2 girls had successful pregnancies; 1 had received radiotherapy
- 1 male developed azoospermia after relapse therapy including alkylating agents
e) Second Neoplasms (SN) (9%)
- 4 cases: breast cancer, pituitary adenoma, osteochondroma, and basal cell carcinoma
- All had received radiotherapy, confirming its role in SN development
f) Musculoskeletal (28%)
- 14% scoliosis, 2% lumbar hyperlordosis
- Often radiation-induced
g) Neurological and Pulmonary Disorders (15% each)
- One case of restrictive lung disease, others with recurrent respiratory infections or congenital anomalies
h) Skin Disorders (36%), Gastrointestinal (17%), Ophthalmologic (15%), and ORL (28%) effects also observed
3. Psychosocial and Quality of Life Impact
- Psychological disorders affected 21% of patients: anxiety, depression, low self-esteem
- 13% experienced academic and social difficulties
- Concerns about fertility were raised by multiple patients during follow-up surveys
- Emphasizes the non-physical burden of long-term survivorship
4. Recurrence and Treatment Intensification
- 11% had relapse, predominantly pulmonary
- 1 patient required intensified chemotherapy due to poor initial response
5. Conclusions and Recommendations
The study confirms that Wilms tumor survivors are at significant risk for multi-system late effects, particularly affecting renal, cardiac, endocrine, and reproductive systems. It calls for:
- Close long-term follow-up
- Minimizing radiotherapy and anthracycline use
- Psychosocial support programs
- Tailored surveillance protocols that include fertility preservation, cardiac screening, and renal monitoring
These efforts are essential to provide survivors with a quality of life comparable to the general population, acknowledging that treatment side effects may appear decades after remission.