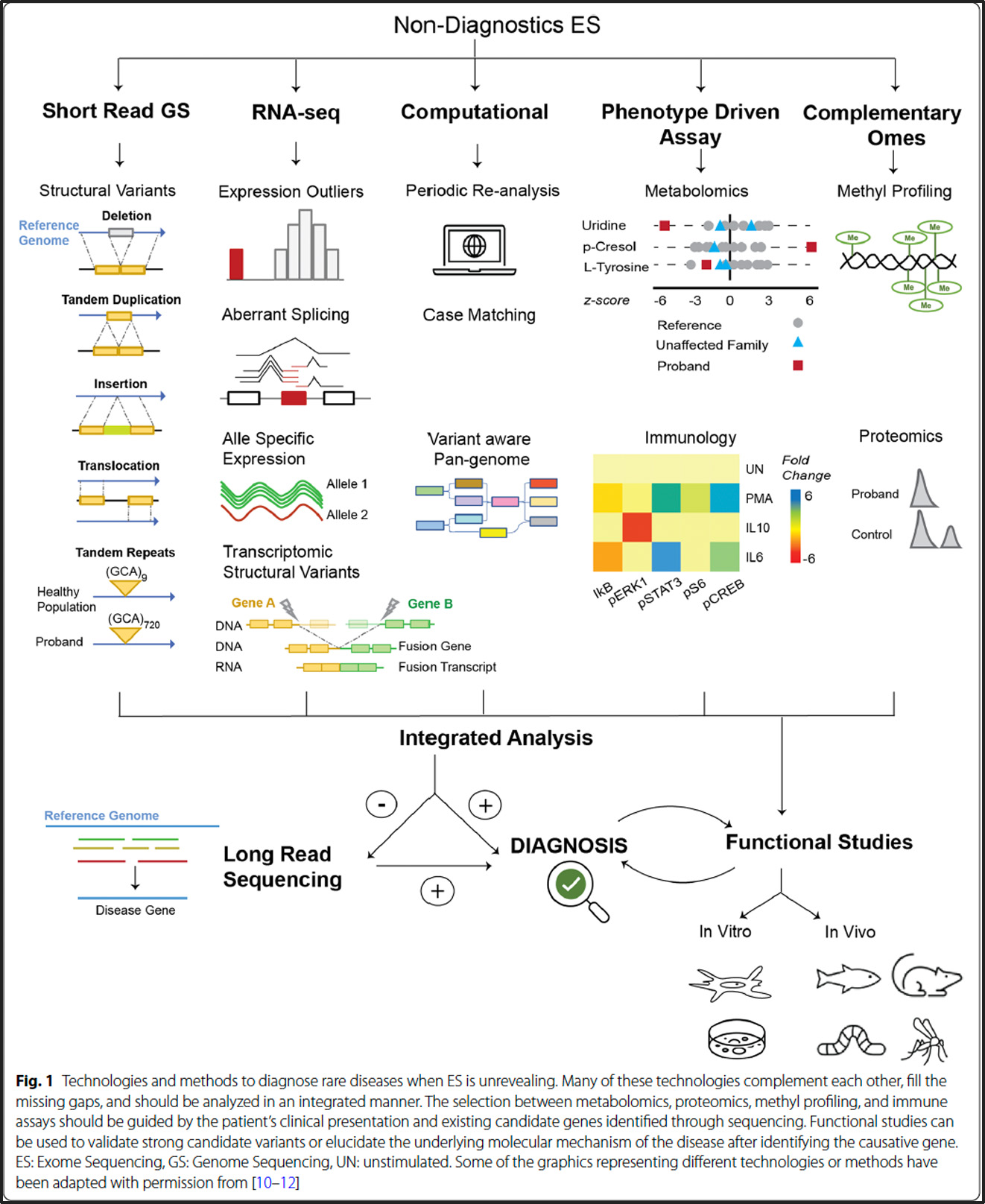
Ces syndromes sont causés respectivement par des mutations avec perte de fonction (LOF) dans les gènes CHD7 et KMT2D, qui codent pour des protéines impliquées dans la régulation épigénétique via le remodelage de la chromatine et les modifications des histones. Bien que distincts, les syndromes de CHARGE et de Kabuki partagent des caractéristiques cliniques telles que des anomalies cardiaques congénitales, une perte auditive, un retard de développement et une déficience intellectuelle, ce qui rend la distinction clinique difficile, en particulier chez les jeunes enfants où les traits faciaux caractéristiques du syndrome de Kabuki ne sont pas toujours évidents.
L’étude part de l’hypothèse que la comparaison des altérations de l’ADNmé à l’échelle du génome chez des individus présentant des mutations LOF hétérozygotes dans CHD7 et KMT2D permettrait d’identifier des signatures d’ADNmé spécifiques à chaque maladie, comprenant à la fois des gènes cibles communs expliquant le chevauchement clinique, et des gènes cibles distincts reflétant les caractéristiques cliniques divergentes.
Méthodologie Pour dériver les signatures d’ADNmé, les chercheurs ont utilisé des échantillons de sang total. La cohorte de Découverte comprenait 19 individus avec le syndrome de CHARGE et des mutations CHD7 LOF (nonsense, frameshift, délétions exoniques, mutations de site d’épissage) et 11 individus avec le syndrome de Kabuki et des mutations KMT2D LOF (nonsense, frameshift). Ces cohortes ont été comparées à des groupes de contrôle appariés en fonction de l’âge et du sexe. Une cohorte de Validation aveugle de 56 échantillons d’ADN avec des variants de CHD7 ou KMT2D (pathogènes, probablement pathogènes, ou de signification inconnue – VUS) a également été utilisée. De plus, 162 échantillons de contrôle d’ADNmé de sang provenant de bases de données publiques (GEO) ont été intégrés pour évaluer la spécificité des signatures.
Les échantillons d’ADN ont été traités par bisulfite de sodium et hybridés sur la puce Illumina Infinium HumanMethylation450 BeadChip Array, qui interroge plus de 480 000 sites CpG. L’analyse statistique a identifié les différences d’ADNmé significatives en utilisant la modélisation de régression limma et le test U de Mann-Whitney non-paramétrique, avec un seuil de signification FDR corrigé de p < 0,01 et une différence moyenne d’ADNmé (Db) supérieure à 10 %. Les sites CpG qui remplissaient ces critères ont été définis comme les signatures d’ADNmé. Des modèles de classification de machine à vecteurs de support (SVM) ont été construits pour prédire la pathogénicité putative des variants. L’indépendance de la composition des types de cellules sanguines a été vérifiée. Les régions différentiellement méthylées (DMR) ont été identifiées à l’aide de la méthode “bump hunting”, et une analyse d’enrichissement fonctionnel a été réalisée pour identifier les voies biologiques. La validation de l’ADNmé a été effectuée par pyroséquençage au bisulfite de sodium pour des loci génomiques sélectionnés.
Résultats
L’étude a identifié des signatures d’ADNmé uniques et hautement spécifiques pour chaque syndrome.
- La signature d’ADNmé CHD7 LOF comprenait 163 sites CpG différentiellement méthylés, distinguant clairement les individus affectés des contrôles.
- La signature d’ADNmé KMT2D LOF comportait 221 sites CpG différentiellement méthylés, permettant également une distinction nette.
Les modèles prédictifs basés sur ces signatures ont démontré une spécificité de 100% : le modèle CHD7 LOF n’a pas classé les individus KMT2D LOF comme positifs pour CHD7, et vice versa. Tous les 162 échantillons de contrôle issus de bases de données publiques ont obtenu de faibles scores de prédiction pour les deux syndromes, confirmant la spécificité des signatures. La validation sur la cohorte aveugle a montré une sensibilité de 100%, classant correctement toutes les mutations CHD7 et KMT2D pathogènes ou probablement pathogènes. Fait intéressant, une mutation pathogène dans KDM6A, un gène également associé au syndrome de Kabuki, a reçu un score élevé du modèle KMT2D LOF, suggérant un chevauchement potentiel des signatures de méthylation entre ces deux gènes.
L’application des modèles prédictifs aux variants de signification inconnue (VUS) a été particulièrement éclairante.
- Sur 13 VUS dans CHD7, 6 ont été classés comme pathogènes et 7 comme bénins par la signature d’ADNmé CHD7 LOF. Ces classifications étaient parfois discordantes avec les critères cliniques existants pour le syndrome de CHARGE (critères de Verloes et Hale) et les outils de prédiction in silico (PolyPhen-2, SIFT, Mutation Taster). Par exemple, trois individus ne répondant pas aux critères cliniques de CHARGE ont été identifiés comme ayant une mutation pathogène par la signature, tandis qu’un individu répondant aux critères n’avait pas la signature.
- Sur 10 VUS dans KMT2D, 1 a été classé comme pathogène et 8 comme bénins. Un VUS a donné un score intermédiaire.
L’analyse des gènes cibles différentiellement méthylés a révélé des chevauchements significatifs. 14 sites CpG étaient partagés par les deux signatures, dont 11 dans le gène HOXA5 et 3 dans SLITRK5.
- Les sites CpG de HOXA5 ont montré un gain d’ADNmé dans les deux signatures CHD7 LOF et KMT2D LOF, un changement validé par pyroséquençage et potentiellement responsable d’une expression réduite de HOXA5.
- Les sites CpG de SLITRK5 ont montré des changements d’ADNmé dans des directions opposées : une perte d’ADNmé dans CHD7 LOF et un gain d’ADNmé dans KMT2D LOF.
Des cibles spécifiques à chaque syndrome ont également été identifiées :
- Pour CHD7 LOF, FOXP2 a montré une perte d’ADNmé.
- Pour KMT2D LOF, MYO1F a montré une perte d’ADNmé.
Les analyses d’enrichissement fonctionnel ont montré que les gènes cibles des signatures d’ADNmé CHD7 LOF et KMT2D LOF sont significativement surreprésentés dans les catégories de processus biologiques liées au développement embryonnaire du cerveau, de l’oreille, du système digestif et d’autres systèmes, reflétant les caractéristiques phénotypiques des syndromes de CHARGE et de Kabuki.
Discussion et Implications
L’étude met en évidence la capacité des signatures d’ADNmé à agir comme des outils moléculaires fonctionnels pour interpréter la pathogénicité des variants de séquence dans CHD7 et KMT2D, offrant une méthode précieuse pour classer les VUS. Ceci est particulièrement pertinent car les critères cliniques et les outils in silico peuvent être inconsistants. La capacité à détecter des mutations LOF qui pourraient ne pas être identifiées par les techniques de séquençage classiques représente une avancée significative pour améliorer les taux de diagnostic moléculaire.
Les résultats confirment également une connexion moléculaire mécanistique entre les syndromes de CHARGE et de Kabuki. L’interaction des protéines CHD7 et KMT2D avec les membres du complexe WAR, déjà suggérée dans des études antérieures, est renforcée par la découverte de cibles CpG communes telles que HOXA5 et SLITRK5. La régulation de HOXA5 par les deux gènes pourrait expliquer des caractéristiques cliniques partagées comme les retards de croissance, les anomalies squelettiques et rénales, et les déficiences du développement neural. De plus, l’implication de FOXP2 dans le syndrome de CHARGE suggère une base moléculaire pour les difficultés de parole et de langage souvent observées.
L’observation que les mutations KDM6A montrent un chevauchement avec la signature KMT2D LOF est également cruciale, car KDM6A est une autre cause du syndrome de Kabuki. Cela suggère que ces deux gènes régulent des ensembles de gènes similaires, ce qui est cohérent avec leurs rôles fonctionnels superposés dans le développement.
L’étude souligne enfin le potentiel des interventions thérapeutiques basées sur des cibles épigénétiques. Des preuves de modèles murins, notamment pour le syndrome de Rett et le syndrome de Kabuki, montrent que les déficits neurologiques peuvent être prévenus ou inversés par des inhibiteurs d’histone désacétylase (HDAC), qui favorisent des états de chromatine ouverte. Cette approche ouvre des perspectives pour de nouvelles thérapies ciblant l’ADNmé dans les troubles neurodéveloppementaux causés par des épigènes.
Conclusion En conclusion, les signatures d’ADNmé spécifiques aux gènes CHD7 et KMT2D offrent une sensibilité et une spécificité élevées, permettant de différencier les mutations pathogènes des variants bénins et de classer les VUS, ce qui est d’une grande valeur clinique. Ces signatures fournissent des preuves de la dysrégulation de gènes clés impliqués dans le développement embryonnaire dans les syndromes de CHARGE et de Kabuki, et mettent en lumière des mécanismes moléculaires partagés et distincts qui sous-tendent leurs caractéristiques cliniques. Leurs applications futures pourraient inclure l’amélioration du diagnostic, la compréhension de la physiopathologie des maladies rares et l’exploration de nouvelles stratégies thérapeutiques ciblant les anomalies épigénétiques. Bien que des validations supplémentaires soient nécessaires, notamment avec des modèles de développement in vitro comme les cellules souches pluripotentes induites (iPSCs), cette recherche représente une avancée prometteuse vers la médecine de précision pour les patients atteints de maladies rares.